%EB%B0%80%EB%8F%84%EB%B2%94%ED%95%A8%EC%88%98
-
 인공지능으로 고성능 양자물성 계산시간 획기적 단축
인공지능과 고성능 과학계산 간의 밀접한 관련성은 최근 2024년도 노벨 물리학상과 화학상이 동시에 수상된 것을 보면 알 수 있다. 우리 연구진이 인공지능을 활용하여 3차원 공간에 분포하는 원자 수준의 화학결합 정보를 예측하여 양자역학적 고성능 컴퓨터 시뮬레이션의 계산 시간을 획기적으로 단축하는데 성공했다.
우리 대학 전기및전자공학부 김용훈 교수팀이 물질의 특성을 도출하기 위해 슈퍼컴퓨터를 활용해 수행되는 원자 수준 양자역학적 계산에 필요한 복잡한 알고리즘을 우회하는 3차원 컴퓨터 비전 인공신경망 기반 계산 방법론을 세계 최초로 개발했다고 30일 밝혔다.
슈퍼컴퓨터를 활용한 양자역학적 밀도범함수론(density functional theory, DFT)* 계산은 빠르면서도 정확하게 양자 물성을 예측할 수 있게 해 첨단 소재 및 약물 설계를 포함한 광범위한 연구·개발 분야에서 표준적인 도구로 자리 잡아 필수 불가결한 역할을 하고 있다.
*밀도범함수론(DFT): 원자 단위에서부터 양자역학적으로 물성을 계산하는 제1원리 계산의 대표적인 이론
그러나 실제 밀도범함수론 계산에서는 3차원적인 전자밀도를 생성한 후 양자역학 방정식을 푸는 복잡한 자기일관장 과정(self-consistent field, SCF)*을 수십에서 수백 번씩 반복해야 해서 그 적용 범위가 수백~수천 개의 원자로 제한되는 한계가 있었다.
*자기일관장(SCF): 상호 연결된 여러 개의 연립 미분 방정식으로 기술해야 하는 복잡한 다체 문제(many-body problem)를 해결하기 위해 널리 사용되는 과학계산법
김용훈 교수 연구팀은 자기일관장 과정을 최근 급속한 발전을 이룬 인공지능 기법으로 회피하는 것이 가능한지 질문했다. 그 결과 3차원 공간에 분포된 화학 결합 정보를 컴퓨터 비전 분야의 신경망 알고리즘을 통해 학습해 계산을 가속화하는 딥SCF(DeepSCF) 모델을 개발했다.
연구진은 밀도범함수론에 따라 전자밀도가 전자들의 양자역학적 정보를 모두 포함하고 있으며 이에 더해 전체 전자밀도와 구성 원자들의 전자밀도의 합 간의 차이인 잔여 전자밀도가 화학결합 정보를 담고 있는 점에 주목하고 기계학습의 목표물로 선정했다.
이후 다양한 화학결합 특성을 포함한 유기 분자들의 데이터 세트를 채택했고 그 안에 포함된 분자들의 원자구조들에 임의의 회전과 변형을 가해 모델의 정확도 및 일반화 성능을 더욱 높였다. 최종적으로 연구팀은 복잡하고 큰 시스템에 대해 딥SCF 방법론의 유효성 및 효율성을 입증했다.
이번 연구를 지도한 김용훈 교수는“3차원 공간에 분포된 양자역학적 화학결합 정보를 인공 신경망에 대응시키는 방법을 찾았다”며 “양자역학적 전자구조 계산이 모든 스케일의 물성 시뮬레이션의 근간이 되므로 인공지능을 통한 물질 계산 가속화의 전반적인 기반 원리를 확립한 것”이라고 연구의 의의를 부여했다.
전기및전자공학부 이룡규 박사과정이 제 1저자로 수행한 이번 연구는 소재 계산 분야의 권위 있는 학술지 '네이쳐 파트너 저널 컴퓨테이셔널 머터리얼즈(Npj Computational Materials)'에 10월 24일 字 온라인판에 게재됐다. (논문명 : Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints)
한편, 이번 연구는 KAIST 석박사 모험사업, 한국연구재단 중견연구자지원사업 등의 지원을 받아 수행되었다.
2024.10.30 조회수 6181
인공지능으로 고성능 양자물성 계산시간 획기적 단축
인공지능과 고성능 과학계산 간의 밀접한 관련성은 최근 2024년도 노벨 물리학상과 화학상이 동시에 수상된 것을 보면 알 수 있다. 우리 연구진이 인공지능을 활용하여 3차원 공간에 분포하는 원자 수준의 화학결합 정보를 예측하여 양자역학적 고성능 컴퓨터 시뮬레이션의 계산 시간을 획기적으로 단축하는데 성공했다.
우리 대학 전기및전자공학부 김용훈 교수팀이 물질의 특성을 도출하기 위해 슈퍼컴퓨터를 활용해 수행되는 원자 수준 양자역학적 계산에 필요한 복잡한 알고리즘을 우회하는 3차원 컴퓨터 비전 인공신경망 기반 계산 방법론을 세계 최초로 개발했다고 30일 밝혔다.
슈퍼컴퓨터를 활용한 양자역학적 밀도범함수론(density functional theory, DFT)* 계산은 빠르면서도 정확하게 양자 물성을 예측할 수 있게 해 첨단 소재 및 약물 설계를 포함한 광범위한 연구·개발 분야에서 표준적인 도구로 자리 잡아 필수 불가결한 역할을 하고 있다.
*밀도범함수론(DFT): 원자 단위에서부터 양자역학적으로 물성을 계산하는 제1원리 계산의 대표적인 이론
그러나 실제 밀도범함수론 계산에서는 3차원적인 전자밀도를 생성한 후 양자역학 방정식을 푸는 복잡한 자기일관장 과정(self-consistent field, SCF)*을 수십에서 수백 번씩 반복해야 해서 그 적용 범위가 수백~수천 개의 원자로 제한되는 한계가 있었다.
*자기일관장(SCF): 상호 연결된 여러 개의 연립 미분 방정식으로 기술해야 하는 복잡한 다체 문제(many-body problem)를 해결하기 위해 널리 사용되는 과학계산법
김용훈 교수 연구팀은 자기일관장 과정을 최근 급속한 발전을 이룬 인공지능 기법으로 회피하는 것이 가능한지 질문했다. 그 결과 3차원 공간에 분포된 화학 결합 정보를 컴퓨터 비전 분야의 신경망 알고리즘을 통해 학습해 계산을 가속화하는 딥SCF(DeepSCF) 모델을 개발했다.
연구진은 밀도범함수론에 따라 전자밀도가 전자들의 양자역학적 정보를 모두 포함하고 있으며 이에 더해 전체 전자밀도와 구성 원자들의 전자밀도의 합 간의 차이인 잔여 전자밀도가 화학결합 정보를 담고 있는 점에 주목하고 기계학습의 목표물로 선정했다.
이후 다양한 화학결합 특성을 포함한 유기 분자들의 데이터 세트를 채택했고 그 안에 포함된 분자들의 원자구조들에 임의의 회전과 변형을 가해 모델의 정확도 및 일반화 성능을 더욱 높였다. 최종적으로 연구팀은 복잡하고 큰 시스템에 대해 딥SCF 방법론의 유효성 및 효율성을 입증했다.
이번 연구를 지도한 김용훈 교수는“3차원 공간에 분포된 양자역학적 화학결합 정보를 인공 신경망에 대응시키는 방법을 찾았다”며 “양자역학적 전자구조 계산이 모든 스케일의 물성 시뮬레이션의 근간이 되므로 인공지능을 통한 물질 계산 가속화의 전반적인 기반 원리를 확립한 것”이라고 연구의 의의를 부여했다.
전기및전자공학부 이룡규 박사과정이 제 1저자로 수행한 이번 연구는 소재 계산 분야의 권위 있는 학술지 '네이쳐 파트너 저널 컴퓨테이셔널 머터리얼즈(Npj Computational Materials)'에 10월 24일 字 온라인판에 게재됐다. (논문명 : Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints)
한편, 이번 연구는 KAIST 석박사 모험사업, 한국연구재단 중견연구자지원사업 등의 지원을 받아 수행되었다.
2024.10.30 조회수 6181 -
 핫전자(뜨거운 전자) 이용, 온실가스 저감원리 규명
우리 연구진이 금속-산화물 계면에서의 촉매 화학 반응 과정에 대한 메커니즘을 직접 밝히고, *핫전자(뜨거운 전자)가 촉매 선택도를 향상시키는 데 결정적인 요소임을 실시간 핫전자 검출을 통해 입증했다.
☞ 핫전자(Hot electron): 분자의 흡착, 화학 촉매 반응, 빛의 흡수와 같은 외부 에너지가 금속 표면에 전달될 때, 화학 에너지의 순간적인 전환과정에서 에너지가 올라간(물질의 자유전자보다 약 100배 높은) 상태의 전자를 말한다. 태양광을 전기에너지로 전환하는 데 사용되는 매개체로도 사용된다.
우리 대학 화학과 박정영 교수(기초과학연구원(IBS) 나노물질 및 화학반응 연구단 부연구단장), 신소재공학과 정연식 교수, 생명화학공학과 정유성 교수 공동연구팀이 차세대 고성능 촉매 설계에 활용할 수 있는 반응성 향상 원리의 기틀을 마련했다고 8일 밝혔다.
현재 에너지 사용량 절감과 친환경 화학 공정 개발이라는 글로벌 도전 과제를 해결하기 위해 새로운 고효율 촉매 소재 개발은 필수적이다. 친환경 화학을 위한 불균일 촉매의 궁극적인 목표는 원하는 생성물에 대해 높은 선택성을 가진 재료를 설계하는 것이다.
많은 화학 촉매 반응 중 알코올의 선택적 산화는 에너지 변환 및 화학 합성에서 중요한 변환 과정이며, 특히 메탄올의 부분 산화를 통해 생성되는 메틸 포르메이트는 포름알데히드 및 포름산과 같은 귀중한 화학 물질의 화학연료로 사용되는 고부가 가치 생성물이다. 따라서 지구온난화 문제의 핵심인 이산화탄소의 저감과 고부가 가치 화학연료 생성의 향상을 위해서는 열역학적인 장벽을 뛰어넘어 높은 선택도를 가지는 우수한 성능의 촉매 개발이 필요하다.
공동연구팀은 이를 해결하는 방안으로, 백금 나노선을 티타늄 산화물에 접합시켜 `금속 나노선-산화물 계면'이 정밀하게 제어되는 신개념의 촉매를 개발했고, 더 나아가 금속 나노선-산화물 계면 기반의 `핫전자 촉매소자'를 활용해 실시간으로 핫전자의 이동을 관찰했다. 연구진은 금속-산화물 계면이 형성되면 메틸 포르메이트의 생성효율이 향상되는 동시에 이산화탄소의 생성이 현저히 감소하는 점을 관찰했으며, 이 높은 선택도는 촉매 표면에 형성된 계면에서의 증폭된 핫전자의 생성과 연관이 있음을 규명했다.
또한 연구진은 계면에서의 증가된 촉매 성능을 실험뿐 아니라 이론적으로도 입증했다. 연구진은 금속 나노선-산화물 계면에서의 증폭된 촉매 선택도가 계면에서의 완전히 다른 촉매 반응 메커니즘에서 기인하는 것임을 양자역학 모델링 계산 결과 비교를 통해 증명했다.
연구를 주도한 화학과 박정영 교수는 "핫전자와 금속 나노선-산화물 계면을 이용해 온실가스인 이산화탄소의 생성을 줄이고 고부가 가치 화학연료의 생성을 증대시킬 수 있다ˮ며 "촉매의 선택도를 핫전자와 금속-산화물 계면을 통해서 제어할 수 있다는 개념은 에너지 전환 및 차세대 촉매 개발에 이용될 수 있고 지구온난화의 주원인인 온실가스의 저감 등의 응용성을 가질 거라고 예상된다ˮ고 말했다.
나노선과 산화물의 접합 연구를 주도한 신소재공학과 정연식 교수는 "기존의 촉매 소자 시스템에서는 기술적으로 어려웠던 금속-산화물 계면에서의 핫전자 검출을 아주 정밀한 나노선 프린팅 기술로 인해 가능하게 만든 연구며, 이 기술은 향후 다양한 차세대 하이브리드 촉매 개발에 활용할 수 있으리라 기대된다ˮ고 말했다.
이론적인 계산으로 계면과 촉매 선택도 간 관계 입증을 주도한 생명화학공학과 정유성 교수 역시 "촉매 화학 반응에서의 선택도를 높이기 위해 금속-산화물 계면이 중요한 역할을 할 수 있음을 실험적 관찰과 이론적인 양자 계산을 통해 증명한 연구로, 불균일 촉매를 이용한 화학 공정 개발에 활용될 수 있을 것으로 기대된다ˮ 라고 언급했다.
우리 대학 화학과 이시우 박사가 제1 저자로 참여하고 기초과학연구원(IBS) 및 한국연구재단의 지원으로 수행된 이번 연구 결과는 종합 과학분야의 국제 학술지 `네이처 커뮤니케이션스(Nature Communications)에 1월 4일 字 게재됐다. (논문명 : Controlling hot electron flux and catalytic selectivity with nanoscale metal-oxide interfaces)
2021.01.11 조회수 67040
핫전자(뜨거운 전자) 이용, 온실가스 저감원리 규명
우리 연구진이 금속-산화물 계면에서의 촉매 화학 반응 과정에 대한 메커니즘을 직접 밝히고, *핫전자(뜨거운 전자)가 촉매 선택도를 향상시키는 데 결정적인 요소임을 실시간 핫전자 검출을 통해 입증했다.
☞ 핫전자(Hot electron): 분자의 흡착, 화학 촉매 반응, 빛의 흡수와 같은 외부 에너지가 금속 표면에 전달될 때, 화학 에너지의 순간적인 전환과정에서 에너지가 올라간(물질의 자유전자보다 약 100배 높은) 상태의 전자를 말한다. 태양광을 전기에너지로 전환하는 데 사용되는 매개체로도 사용된다.
우리 대학 화학과 박정영 교수(기초과학연구원(IBS) 나노물질 및 화학반응 연구단 부연구단장), 신소재공학과 정연식 교수, 생명화학공학과 정유성 교수 공동연구팀이 차세대 고성능 촉매 설계에 활용할 수 있는 반응성 향상 원리의 기틀을 마련했다고 8일 밝혔다.
현재 에너지 사용량 절감과 친환경 화학 공정 개발이라는 글로벌 도전 과제를 해결하기 위해 새로운 고효율 촉매 소재 개발은 필수적이다. 친환경 화학을 위한 불균일 촉매의 궁극적인 목표는 원하는 생성물에 대해 높은 선택성을 가진 재료를 설계하는 것이다.
많은 화학 촉매 반응 중 알코올의 선택적 산화는 에너지 변환 및 화학 합성에서 중요한 변환 과정이며, 특히 메탄올의 부분 산화를 통해 생성되는 메틸 포르메이트는 포름알데히드 및 포름산과 같은 귀중한 화학 물질의 화학연료로 사용되는 고부가 가치 생성물이다. 따라서 지구온난화 문제의 핵심인 이산화탄소의 저감과 고부가 가치 화학연료 생성의 향상을 위해서는 열역학적인 장벽을 뛰어넘어 높은 선택도를 가지는 우수한 성능의 촉매 개발이 필요하다.
공동연구팀은 이를 해결하는 방안으로, 백금 나노선을 티타늄 산화물에 접합시켜 `금속 나노선-산화물 계면'이 정밀하게 제어되는 신개념의 촉매를 개발했고, 더 나아가 금속 나노선-산화물 계면 기반의 `핫전자 촉매소자'를 활용해 실시간으로 핫전자의 이동을 관찰했다. 연구진은 금속-산화물 계면이 형성되면 메틸 포르메이트의 생성효율이 향상되는 동시에 이산화탄소의 생성이 현저히 감소하는 점을 관찰했으며, 이 높은 선택도는 촉매 표면에 형성된 계면에서의 증폭된 핫전자의 생성과 연관이 있음을 규명했다.
또한 연구진은 계면에서의 증가된 촉매 성능을 실험뿐 아니라 이론적으로도 입증했다. 연구진은 금속 나노선-산화물 계면에서의 증폭된 촉매 선택도가 계면에서의 완전히 다른 촉매 반응 메커니즘에서 기인하는 것임을 양자역학 모델링 계산 결과 비교를 통해 증명했다.
연구를 주도한 화학과 박정영 교수는 "핫전자와 금속 나노선-산화물 계면을 이용해 온실가스인 이산화탄소의 생성을 줄이고 고부가 가치 화학연료의 생성을 증대시킬 수 있다ˮ며 "촉매의 선택도를 핫전자와 금속-산화물 계면을 통해서 제어할 수 있다는 개념은 에너지 전환 및 차세대 촉매 개발에 이용될 수 있고 지구온난화의 주원인인 온실가스의 저감 등의 응용성을 가질 거라고 예상된다ˮ고 말했다.
나노선과 산화물의 접합 연구를 주도한 신소재공학과 정연식 교수는 "기존의 촉매 소자 시스템에서는 기술적으로 어려웠던 금속-산화물 계면에서의 핫전자 검출을 아주 정밀한 나노선 프린팅 기술로 인해 가능하게 만든 연구며, 이 기술은 향후 다양한 차세대 하이브리드 촉매 개발에 활용할 수 있으리라 기대된다ˮ고 말했다.
이론적인 계산으로 계면과 촉매 선택도 간 관계 입증을 주도한 생명화학공학과 정유성 교수 역시 "촉매 화학 반응에서의 선택도를 높이기 위해 금속-산화물 계면이 중요한 역할을 할 수 있음을 실험적 관찰과 이론적인 양자 계산을 통해 증명한 연구로, 불균일 촉매를 이용한 화학 공정 개발에 활용될 수 있을 것으로 기대된다ˮ 라고 언급했다.
우리 대학 화학과 이시우 박사가 제1 저자로 참여하고 기초과학연구원(IBS) 및 한국연구재단의 지원으로 수행된 이번 연구 결과는 종합 과학분야의 국제 학술지 `네이처 커뮤니케이션스(Nature Communications)에 1월 4일 字 게재됐다. (논문명 : Controlling hot electron flux and catalytic selectivity with nanoscale metal-oxide interfaces)
2021.01.11 조회수 67040 -
 오차율 10% 이내 정확도의 소재 설계 기술 개발
우리 대학 화학과 김형준 교수 연구팀이 소재 물성의 예측 오차율을 기존 기술보다 30% 이상 줄여 정확도를 한층 높인 소재 시뮬레이션 설계 기술을 개발했다.
이번 기술 개발을 통해 기존 40%에 달했던 소재 물성 예측 오차율을 10% 내로 줄임으로써 소재 개발에 걸리는 시간과 비용을 크게 절약할 수 있을 것으로 기대된다.
김민호 박사와 창원대 김원준 교수가 공동 1 저자로 참여한 이번 연구 결과는 국제 학술지 ‘미국 화학회지(Journal of the American Chemical Societry)’ 1월 10일 자 온라인판에 게재됐다. (논문명 : uMBD: A Materials-Ready Dispersion Correction that Uniformly Treats Metallic, Ionic, and van der Waals Bonding)
새로운 기능성 소재 개발의 중요성이 커지면서 컴퓨터 시뮬레이션을 이용해 소재 물성을 정확히 예측해 새로운 소재를 설계하는 기술이 주목받고 있다.
소재 시뮬레이션 기술은 실제로 소재를 합성하고 평가하기 전에 가상 실험으로 다양한 소재 물성을 예측 및 설계하는 기술로, 주로 밀도범함수 이론(Density functional theory)이라는 양자 이론에 바탕을 두고 있다.
기존의 밀도범함수 이론은 소재 계면에서 반데르발스 힘을 정확하게 설명하지 못한다는 문제가 있었다. 반데르발스 힘은 전하의 일시적 쏠림으로 인해 분자가 순간적으로 극성을 띠면서 나타나는 당기는 힘을 뜻하는데, 이를 정확히 기술하지 못하기 때문에 소재 물성 예측 정확도가 떨어진다는 한계가 있다.
연구팀은 반데르발스 힘을 정확하고 효과적으로 기술할 수 있는 새로운 이론을 개발하고, 이를 밀도범함수 이론에 접목해 소재 시뮬레이션 기술의 정확도를 한층 높이는 데 성공했다.
연구팀은 100여 종의 다양한 소재를 테스트한 결과 40% 정도에 달했던 기존의 소재 물성 예측 오차율이 새 기술을 통해 10% 이내로 줄어듦을 확인했다.
특히 반데르발스 힘은 분자 소재부터 금속 및 반도체 소재에 이르기까지 거의 모든 재료 내에서 소재 물성을 결정하는 데 중요한 역할을 해, 연구팀의 새로운 이론은 다양한 차세대 기능성 소재 설계 연구에 적용 가능할 것으로 기대된다.
실제로 연구팀의 새 시뮬레이션 방법을 통해 리튬 이온 배터리 물질의 전압이나 2차원 소재의 박리 에너지를 예측하는 과정에서 높은 정확도를 보인 것으로 확인됐다.
김형준 교수는 “소재 개발 연구에 있어 경쟁력 강화를 위해서 기초 연구의 중요성이 점차 커지고 있다”라며 “새로 개발한 소재 시뮬레이션 기술을 배터리 소재, 에너지 전환 촉매 소재, 2차원 나노 소재 등 다양한 기능성 소재 설계 연구에 적용할 수 있을 것이다”라고 말했다.
이번 연구는 한국연구재단의 미래소재디스커버리 사업과 선도연구센터 지원 사업 (SRC)의 지원을 통해 수행됐다.
□ 그림 설명
그림1. 새롭게 개발한 이론 (uMBD)을 이용한 소재 시뮬레이션 기술과 기능성 소재 설계
2020.01.29 조회수 14792
오차율 10% 이내 정확도의 소재 설계 기술 개발
우리 대학 화학과 김형준 교수 연구팀이 소재 물성의 예측 오차율을 기존 기술보다 30% 이상 줄여 정확도를 한층 높인 소재 시뮬레이션 설계 기술을 개발했다.
이번 기술 개발을 통해 기존 40%에 달했던 소재 물성 예측 오차율을 10% 내로 줄임으로써 소재 개발에 걸리는 시간과 비용을 크게 절약할 수 있을 것으로 기대된다.
김민호 박사와 창원대 김원준 교수가 공동 1 저자로 참여한 이번 연구 결과는 국제 학술지 ‘미국 화학회지(Journal of the American Chemical Societry)’ 1월 10일 자 온라인판에 게재됐다. (논문명 : uMBD: A Materials-Ready Dispersion Correction that Uniformly Treats Metallic, Ionic, and van der Waals Bonding)
새로운 기능성 소재 개발의 중요성이 커지면서 컴퓨터 시뮬레이션을 이용해 소재 물성을 정확히 예측해 새로운 소재를 설계하는 기술이 주목받고 있다.
소재 시뮬레이션 기술은 실제로 소재를 합성하고 평가하기 전에 가상 실험으로 다양한 소재 물성을 예측 및 설계하는 기술로, 주로 밀도범함수 이론(Density functional theory)이라는 양자 이론에 바탕을 두고 있다.
기존의 밀도범함수 이론은 소재 계면에서 반데르발스 힘을 정확하게 설명하지 못한다는 문제가 있었다. 반데르발스 힘은 전하의 일시적 쏠림으로 인해 분자가 순간적으로 극성을 띠면서 나타나는 당기는 힘을 뜻하는데, 이를 정확히 기술하지 못하기 때문에 소재 물성 예측 정확도가 떨어진다는 한계가 있다.
연구팀은 반데르발스 힘을 정확하고 효과적으로 기술할 수 있는 새로운 이론을 개발하고, 이를 밀도범함수 이론에 접목해 소재 시뮬레이션 기술의 정확도를 한층 높이는 데 성공했다.
연구팀은 100여 종의 다양한 소재를 테스트한 결과 40% 정도에 달했던 기존의 소재 물성 예측 오차율이 새 기술을 통해 10% 이내로 줄어듦을 확인했다.
특히 반데르발스 힘은 분자 소재부터 금속 및 반도체 소재에 이르기까지 거의 모든 재료 내에서 소재 물성을 결정하는 데 중요한 역할을 해, 연구팀의 새로운 이론은 다양한 차세대 기능성 소재 설계 연구에 적용 가능할 것으로 기대된다.
실제로 연구팀의 새 시뮬레이션 방법을 통해 리튬 이온 배터리 물질의 전압이나 2차원 소재의 박리 에너지를 예측하는 과정에서 높은 정확도를 보인 것으로 확인됐다.
김형준 교수는 “소재 개발 연구에 있어 경쟁력 강화를 위해서 기초 연구의 중요성이 점차 커지고 있다”라며 “새로 개발한 소재 시뮬레이션 기술을 배터리 소재, 에너지 전환 촉매 소재, 2차원 나노 소재 등 다양한 기능성 소재 설계 연구에 적용할 수 있을 것이다”라고 말했다.
이번 연구는 한국연구재단의 미래소재디스커버리 사업과 선도연구센터 지원 사업 (SRC)의 지원을 통해 수행됐다.
□ 그림 설명
그림1. 새롭게 개발한 이론 (uMBD)을 이용한 소재 시뮬레이션 기술과 기능성 소재 설계
2020.01.29 조회수 14792 -
 박정영, 정유성 교수, 합금 나노촉매 성능 향상 원리 밝혀
〈 박 정 영 교수, 정 유 성 교수〉
우리 대학 EEWS 대학원 및 화학과 박정영 교수 연구팀이 정유성 교수 연구팀과의 공동 연구를 통해 합금 나노 촉매 표면에 형성된 금속-산화물 계면이 촉매 성능을 향상시키는 중요한 요소임을 밝혔다.
이번 연구결과는 종합 과학 분야 국제 학술지 ‘네이처 커뮤니케이션즈’(Nature Communications) 6월 8일자 온라인 판에 게재됐다.
합금 나노입자는 높은 효율의 촉매 활성도를 가져 석유화학 공정뿐만 아니라 수소 연료 전지, 물 분해 등 친환경 촉매로 주목받고 있다. 합금 나노입자는 화학적 조성에 따라 촉매 표면의 전자 구조 및 결합 에너지를 제어할 수 있어 활용성이 크다.
이런 우수한 특성에도 불구하고 실제 촉매 환경에서는 반응물과 조건에 따라 나노 입자 표면 구조가 쉽게 달라져 합금 나노 촉매의 반응 원리 규명에 어려움이 있었다.
촉매 반응의 원리를 결정하는 핵심 요소는 핫전자이다. 화학반응이 일어날 때 촉매 표면에 순간적으로(펨토초, 1천조분의 1초) 발생하고 사라지지만 촉매 반응의 활성도를 파악할 수 있는 척도와 같다. 촉매 활성도가 증가하면 핫전자 양도 늘어나기 때문이다. 실시간으로 핫전자를 직접 검출할 수 있는 방법이 마땅히 없던 중 2015년 박정영 부연구단장 연구진이 핫전자를 관찰할 수 있는 핫전자 촉매센서를 개발했다. 이후 박 부연구단장은 핫전자 촉매센서를 중심으로 활발한 연구를 통해 다양한 결과를 내고 있다.
이번 연구에서는 백금과 코발트가 합금된 나노입자를 핫전자 촉매센서에 접목하는 방식으로 연구를 설계했다. 백금-코발트 합금 나노입자는 화학산업 및 에너지·환경 분야에 중요한 촉매 구성요소다. 백금-코발트 합금 나노입자처럼 복잡한 구조를 가진 나노 촉매 구조에 핫전자 촉매센서를 적용해 실시간으로 핫전자를 관찰하는 것이 이번 실험의 큰 관건이었다.
먼저 연구진은 여러 비율로 백금과 코발트를 합성해 합금 나노 촉매들을 제작하고 핫전자 촉매센서를 적용했다. 그 결과 75% 백금과 25% 코발트 비율로 합금 나노입자를 합성할 경우, 가장 많은 핫전자가 발생하고 촉매 성능이 높다는 것을 확인했다. 이후 핫전자 발생량과 촉매 성능의 상관관계를 보다 명확히 밝히고자 실시간 투과전자현미경(TEM, Transmission Electron Microscopy)으로 실험 과정을 관찰했다.
수소산화 반응에 합금 나노촉매를 적용하자 한 층의 코발트 산화물이 백금-코발트 합금 나노 입자 표면 위에 형성되면서 금속-산화물(백금-코발트 산화물)계면이 만들어졌다. 금속-산화물 계면에서 전하 이동이 늘어나면서 핫전자 검출 효율이 증가한 것이다. 다시 말해 금속-산화물 계면이 합금 나노 촉매의 활성을 높이는 데 결정적임을 실제로 입증한 것이다.
이번 연구는 실험 뿐 아니라 이론적으로도 계면과 촉매 성능 간 상관관계를 입증했다. 정유성 교수 연구진은 밀도범함수이론(Density Functional Theory) 기반의 양자계산을 통해 백금-코발트 산화물 계면에서 낮은 활성화 에너지로 일어나는 반응 원리를 이론적으로 뒷받침해 핫전자 발생 및 촉매 성능에 대한 근원적인 해석을 제안했다.
정 교수는 “이번 결과는 촉매 연구자들이 금속-산화물 계면의 중요성을 다시 주목하게 되는 계기가 될 것이다”고 말했다.
박 교수는 “이번 연구로 합금 나노촉매의 반응 중 자연스럽게 형성되는 두 물질 사이의 계면이 촉매 반응성과 핫전자의 생성을 증폭시킨다는 점을 규명했다”며 “실제 촉매반응이 일어나는 상압과 고온 환경에서 얻어진 결과를 토대로 향후 고효율의 차세대 촉매물질을 개발하는데 연구 결과를 응용할 수 있다”고 전망했다.
□ 그림 설명
그림1. 나노 촉매계면에서의 핫전자 움직임 실시간 관찰
그림2. 핫전자 촉매센서를 이용한 합금 나노입자에서의 핫전자 움직임 관찰
2018.07.06 조회수 14067
박정영, 정유성 교수, 합금 나노촉매 성능 향상 원리 밝혀
〈 박 정 영 교수, 정 유 성 교수〉
우리 대학 EEWS 대학원 및 화학과 박정영 교수 연구팀이 정유성 교수 연구팀과의 공동 연구를 통해 합금 나노 촉매 표면에 형성된 금속-산화물 계면이 촉매 성능을 향상시키는 중요한 요소임을 밝혔다.
이번 연구결과는 종합 과학 분야 국제 학술지 ‘네이처 커뮤니케이션즈’(Nature Communications) 6월 8일자 온라인 판에 게재됐다.
합금 나노입자는 높은 효율의 촉매 활성도를 가져 석유화학 공정뿐만 아니라 수소 연료 전지, 물 분해 등 친환경 촉매로 주목받고 있다. 합금 나노입자는 화학적 조성에 따라 촉매 표면의 전자 구조 및 결합 에너지를 제어할 수 있어 활용성이 크다.
이런 우수한 특성에도 불구하고 실제 촉매 환경에서는 반응물과 조건에 따라 나노 입자 표면 구조가 쉽게 달라져 합금 나노 촉매의 반응 원리 규명에 어려움이 있었다.
촉매 반응의 원리를 결정하는 핵심 요소는 핫전자이다. 화학반응이 일어날 때 촉매 표면에 순간적으로(펨토초, 1천조분의 1초) 발생하고 사라지지만 촉매 반응의 활성도를 파악할 수 있는 척도와 같다. 촉매 활성도가 증가하면 핫전자 양도 늘어나기 때문이다. 실시간으로 핫전자를 직접 검출할 수 있는 방법이 마땅히 없던 중 2015년 박정영 부연구단장 연구진이 핫전자를 관찰할 수 있는 핫전자 촉매센서를 개발했다. 이후 박 부연구단장은 핫전자 촉매센서를 중심으로 활발한 연구를 통해 다양한 결과를 내고 있다.
이번 연구에서는 백금과 코발트가 합금된 나노입자를 핫전자 촉매센서에 접목하는 방식으로 연구를 설계했다. 백금-코발트 합금 나노입자는 화학산업 및 에너지·환경 분야에 중요한 촉매 구성요소다. 백금-코발트 합금 나노입자처럼 복잡한 구조를 가진 나노 촉매 구조에 핫전자 촉매센서를 적용해 실시간으로 핫전자를 관찰하는 것이 이번 실험의 큰 관건이었다.
먼저 연구진은 여러 비율로 백금과 코발트를 합성해 합금 나노 촉매들을 제작하고 핫전자 촉매센서를 적용했다. 그 결과 75% 백금과 25% 코발트 비율로 합금 나노입자를 합성할 경우, 가장 많은 핫전자가 발생하고 촉매 성능이 높다는 것을 확인했다. 이후 핫전자 발생량과 촉매 성능의 상관관계를 보다 명확히 밝히고자 실시간 투과전자현미경(TEM, Transmission Electron Microscopy)으로 실험 과정을 관찰했다.
수소산화 반응에 합금 나노촉매를 적용하자 한 층의 코발트 산화물이 백금-코발트 합금 나노 입자 표면 위에 형성되면서 금속-산화물(백금-코발트 산화물)계면이 만들어졌다. 금속-산화물 계면에서 전하 이동이 늘어나면서 핫전자 검출 효율이 증가한 것이다. 다시 말해 금속-산화물 계면이 합금 나노 촉매의 활성을 높이는 데 결정적임을 실제로 입증한 것이다.
이번 연구는 실험 뿐 아니라 이론적으로도 계면과 촉매 성능 간 상관관계를 입증했다. 정유성 교수 연구진은 밀도범함수이론(Density Functional Theory) 기반의 양자계산을 통해 백금-코발트 산화물 계면에서 낮은 활성화 에너지로 일어나는 반응 원리를 이론적으로 뒷받침해 핫전자 발생 및 촉매 성능에 대한 근원적인 해석을 제안했다.
정 교수는 “이번 결과는 촉매 연구자들이 금속-산화물 계면의 중요성을 다시 주목하게 되는 계기가 될 것이다”고 말했다.
박 교수는 “이번 연구로 합금 나노촉매의 반응 중 자연스럽게 형성되는 두 물질 사이의 계면이 촉매 반응성과 핫전자의 생성을 증폭시킨다는 점을 규명했다”며 “실제 촉매반응이 일어나는 상압과 고온 환경에서 얻어진 결과를 토대로 향후 고효율의 차세대 촉매물질을 개발하는데 연구 결과를 응용할 수 있다”고 전망했다.
□ 그림 설명
그림1. 나노 촉매계면에서의 핫전자 움직임 실시간 관찰
그림2. 핫전자 촉매센서를 이용한 합금 나노입자에서의 핫전자 움직임 관찰
2018.07.06 조회수 14067 -
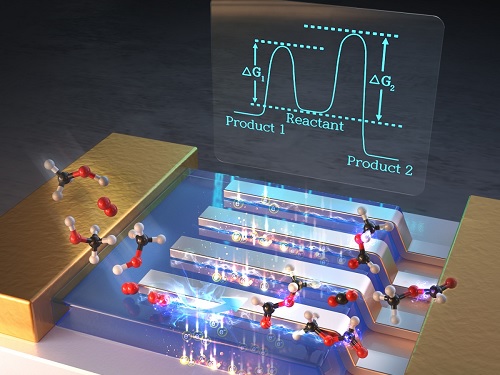
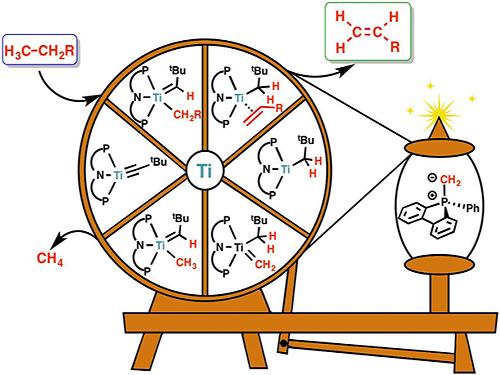 백무현 교수, 타이타늄 촉매반응으로 화학소재 올레핀 합성 성공
〈 백 무 현 교수 〉
우리 대학 화학과 백무현 교수 연구팀이 우리 주변에 흔한 타이타늄(Titanium) 촉매를 활용해 플라스틱, 의약품 원료로 사용하는 올레핀(olefins) 합성에 성공했다.
석유화학산업 분야 주요 소재인 올레핀은 보통 800℃ 고온으로 석유를 증기 분해(steam cracking)해 제조한다. 매우 높은 열과 에너지가 투입되고 이산화탄소 등 온실가스가 발생하는 것이 단점이다.
연구결과는 27일 국제학술지 네이처 케미스트리에 게재됐다.
기초과학연구원 분자활성 촉매반응 연구단의 부연구단장으로 재직 중인 백무현 교수는 계산화학을 통해 타이타늄을 최적의 촉매로 선택했고 탄화수소(hydrocarbon)의 수소를 선택적으로 없애는 탈수소반응을 구현했다. 이로써 기존 공정에 비해 10분의 1정도 낮은 온도(75℃)에서 올레핀을 합성했다.
올레핀은 플라스틱, 고분자 화합물, 의약품 등에 활용하는 기초 원료이다. 활용도가 커 올레핀 합성 과정은 많은 연구자들이 연구주제로 삼고 있다.
올레핀은 탄화수소가 수소를 잃으면서 탄소(C) 두 개가 이중결합(C=C)해 생성되는데 증기 분해 방식은 반응 중 탄소-탄소 결합이 끊어져 올레핀 혼합물이나 다른 탄화수소들이 합성되는 단점이 있다. 또 석유 대신 천연가스에서 올레핀을 합성하려면 온실가스가 발생해 오염과 공해 문제가 뒤따랐다.
화학자들은 석유와 천연가스 등 탄화수소 화합물을 가공하거나 분해할 때 열과 에너지를 적게 사용하고, 환경오염이 덜한 화학반응을 구현하기 위해 다양한 촉매반응을 연구했다.
탄소와 수소만으로 결합된 탄화수소는 두 분자 간 결합이 매우 강하기 때문에 결합을 끊고 반응을 유도하는 촉매 개발이 주요 과제였다. 이리듐(Iridium), 로듐(rhodium), 루테늄(ruthenium) 등 전이금속을 촉매로 적용했으나 비용이 너무 비싸 실제 산업에 활용하기는 어려웠다.
백 부단장은 비싼 전이금속 보다 수십 배 저렴한 타이타늄을 촉매로 적용했다. 백 부단장은 밀도범함수를 활용한 계산 화학을 통해 최적의 촉매 후보물질로 타이타늄을 제안했고 미국 펜실베니아대학 연구진은 약 75℃에서 탈수소반응이 성공적으로 이뤄졌음을 실험으로 확인했다.
지난해 이리듐 촉매로 메탄가스의 강력한 탄소-수소 결합을 분해한 데 이어 이번 연구에서도 계산화학으로 정확한 촉매를 예측했다. 또 탈수소반응에 이리듐 촉매를 활용할 때 탄화수소가 이성질화(isomerization) 되는 문제도 타이타늄 촉매로 해결됨을 관찰했다.
백 교수는 “이리듐은 반응성이 매우 크지만 값이 비싸고 구하기 어렵다. 반면 타이타늄은 값이 매우 저렴하고 구하기 쉽다”며 “향후 타이타늄 촉매의 반응성과 효율성을 높인다면 기존 올레핀 합성공정의 비용이 줄어들 것”이라고 말했다.
이번 연구는 미국 펜실베니아 대학의 대니얼 민디올라(Daniel J. Mindiola) 교수 그룹과 공동으로 진행됐다.
□ 그림 설명
그림1. 연구진이 제안한 타이타늄 촉매를 활용한 탈수소반응 메커니즘
그림2. 밀도범함수를 활용한 계산화학으로 본 탈수소반응 메커니즘
2017.06.28 조회수 21227
백무현 교수, 타이타늄 촉매반응으로 화학소재 올레핀 합성 성공
〈 백 무 현 교수 〉
우리 대학 화학과 백무현 교수 연구팀이 우리 주변에 흔한 타이타늄(Titanium) 촉매를 활용해 플라스틱, 의약품 원료로 사용하는 올레핀(olefins) 합성에 성공했다.
석유화학산업 분야 주요 소재인 올레핀은 보통 800℃ 고온으로 석유를 증기 분해(steam cracking)해 제조한다. 매우 높은 열과 에너지가 투입되고 이산화탄소 등 온실가스가 발생하는 것이 단점이다.
연구결과는 27일 국제학술지 네이처 케미스트리에 게재됐다.
기초과학연구원 분자활성 촉매반응 연구단의 부연구단장으로 재직 중인 백무현 교수는 계산화학을 통해 타이타늄을 최적의 촉매로 선택했고 탄화수소(hydrocarbon)의 수소를 선택적으로 없애는 탈수소반응을 구현했다. 이로써 기존 공정에 비해 10분의 1정도 낮은 온도(75℃)에서 올레핀을 합성했다.
올레핀은 플라스틱, 고분자 화합물, 의약품 등에 활용하는 기초 원료이다. 활용도가 커 올레핀 합성 과정은 많은 연구자들이 연구주제로 삼고 있다.
올레핀은 탄화수소가 수소를 잃으면서 탄소(C) 두 개가 이중결합(C=C)해 생성되는데 증기 분해 방식은 반응 중 탄소-탄소 결합이 끊어져 올레핀 혼합물이나 다른 탄화수소들이 합성되는 단점이 있다. 또 석유 대신 천연가스에서 올레핀을 합성하려면 온실가스가 발생해 오염과 공해 문제가 뒤따랐다.
화학자들은 석유와 천연가스 등 탄화수소 화합물을 가공하거나 분해할 때 열과 에너지를 적게 사용하고, 환경오염이 덜한 화학반응을 구현하기 위해 다양한 촉매반응을 연구했다.
탄소와 수소만으로 결합된 탄화수소는 두 분자 간 결합이 매우 강하기 때문에 결합을 끊고 반응을 유도하는 촉매 개발이 주요 과제였다. 이리듐(Iridium), 로듐(rhodium), 루테늄(ruthenium) 등 전이금속을 촉매로 적용했으나 비용이 너무 비싸 실제 산업에 활용하기는 어려웠다.
백 부단장은 비싼 전이금속 보다 수십 배 저렴한 타이타늄을 촉매로 적용했다. 백 부단장은 밀도범함수를 활용한 계산 화학을 통해 최적의 촉매 후보물질로 타이타늄을 제안했고 미국 펜실베니아대학 연구진은 약 75℃에서 탈수소반응이 성공적으로 이뤄졌음을 실험으로 확인했다.
지난해 이리듐 촉매로 메탄가스의 강력한 탄소-수소 결합을 분해한 데 이어 이번 연구에서도 계산화학으로 정확한 촉매를 예측했다. 또 탈수소반응에 이리듐 촉매를 활용할 때 탄화수소가 이성질화(isomerization) 되는 문제도 타이타늄 촉매로 해결됨을 관찰했다.
백 교수는 “이리듐은 반응성이 매우 크지만 값이 비싸고 구하기 어렵다. 반면 타이타늄은 값이 매우 저렴하고 구하기 쉽다”며 “향후 타이타늄 촉매의 반응성과 효율성을 높인다면 기존 올레핀 합성공정의 비용이 줄어들 것”이라고 말했다.
이번 연구는 미국 펜실베니아 대학의 대니얼 민디올라(Daniel J. Mindiola) 교수 그룹과 공동으로 진행됐다.
□ 그림 설명
그림1. 연구진이 제안한 타이타늄 촉매를 활용한 탈수소반응 메커니즘
그림2. 밀도범함수를 활용한 계산화학으로 본 탈수소반응 메커니즘
2017.06.28 조회수 21227 -
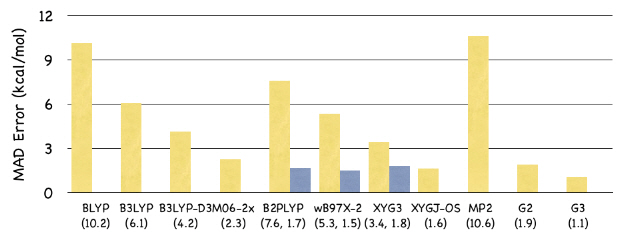 실험비용 획기적으로 절감하는 빠르고 정확한 양자역학 계산 이론 개발
정유성 교수 윌리엄 고다드 교수
현재 널리 사용되고 있는 양자역학 원리를 이용하여 정확하면서도 계산시간이 획기적으로 개선된 새로운 전자밀도범함수 계산이론*이 국내 연구진에 의해 개발됐다.
*) 전자밀도범함수 계산이론 : 비교적 간단한 파동함수와 전자밀도만으로 에너지와 성질을 계산할 수 있음을 증명한 이론
우리 학교 EEWS대학원 정유성 교수(38세)와 윌리엄 고다드 교수가 주도한 이번 연구는 교육과학기술부(장관 이주호)와 한국연구재단이 추진하는 WCU(세계수준의 연구중심대학)육성사업의 지원을 받아 수행되었고, 연구결과는 자연과학분야의 권위 있는 학술지인 ‘미국립과학원회보(PNAS)’ 11월 23일자 온라인으로 게재되었다.
정유성 교수와 고다드 교수는 기존의 양자계산의 문제점인 계산시간과 부정확한 예측으로 인한 결과의 신뢰성이 떨어지는 단점을 보완하여, 정확하면서도 빠른 전자밀도범함수 이론과 알고리즘을 개발하였다.
양자역학 계산법 중 파동함수*를 이용하면 정확도가 높은 반면에 계산시간도 빠르게 증가해 수백-수천 개의 원자를 갖는 거대 분자에 적용하기 어렵고, 상대적으로 계산량이 적은 전자밀도를 변수로 사용할 경우 적용할 수 있는 분자의 크기는 증가하지만 정확도가 떨어지는 단점이 있었다.
*) 파동함수(波動函數) : 양자역학에서 물질입자인 전자·양성자·중성자 등의 상태를 나타내는 양
전자들의 상호작용은 스핀*이 같은 전자들끼리의 상호작용과 스핀이 다른 전자들끼리의 상호작용으로 나뉘는데, 파울리의 배타 원리에 의해 스핀이 다른 전자 사이의 거리가 더 가까우므로 스핀이 다른 전자들의 상호작용이 더 크다. 연구팀은 이 점에 착안하여, 기존에 존재하던 정확한 계산법에서 스핀이 다른 전자 사이의 상호작용을 중점적으로 계산하여 속도를 향상시켰다.
*) 스핀(spin) : 입자의 기본성질을 나타내는 물리량 중 하나로, 입자의 고유한 운동량을 나타냄. 소립자들의 특징을 밝히는 중요한 물리량임
또한 전자들의 상호작용은 국소성을 띠기 때문에 멀리 떨어진 전자 사이에는 상호작용이 거의 없어서 이들을 무시하더라도 총 에너지에 영향을 미치지 않는다는 점에 착안하여, 계산시간을 최대 100배이상 단축시키는 알고리즘을 개발하였다. 예를 들어, 기존의 계산방법으로는 탄소 200개와 수소 402개로 이루어진 알케인(aklane) 분자를 정확히 계산하는데 6개월이 걸린 반면, 새로운 방법론을 이용하면 비슷한 정확도로 하루(24시간)면 계산할 수 있다.
정유성 교수는 “그동안 국내의 계산과학 및 재료 설계 커뮤니티가 응용 연구에 주로 집중하여 짧은 시간 동안 훌륭한 결과를 많이 도출한 반면, 상대적으로 긴 시간을 요구하는 기초 방법론이나 소프트웨어 개발에서는 국제경쟁력이 뒤처져 있는 추세였다. 그러나 이번 연구는 기존의 방법들보다 월등한 정확도와 속도를 가진 방법론을 국내에서 개발했다는 점에서 의미가 크다”고 연구의의를 밝혔다.
아울러 이번에 개발된 방법론은 큐켐(Q-CHEM)이라는 상용 소프트웨어 패키지를 통해 일반연구자들에게 공급될 예정이다.알케인(alkane) 분자의 크기에 따라 본 연구에서 제시한 새로운 방법론(local XYGJ-OS)과 기존의 방법론의 계산 시간을 비교한 그래프. local XYGJ-OS는 전자 간 상호작용의 국소성을 이용해 계산 시간을 낮춘 방법이다.다양한 양자계산 방법과 본 연구에서 제시한 XYGJ-OS 방법의 오차. 233개 분자의 생성열을 실험값과 대조하여 오차의 절대값을 평균하였다. B2PLYP부터 XYG3까지의 방법 및 G2, G3 방법은 XYGJ-OS에 비하여 훨씬 많은 시간을 소모한다.
2011.11.29 조회수 17989
실험비용 획기적으로 절감하는 빠르고 정확한 양자역학 계산 이론 개발
정유성 교수 윌리엄 고다드 교수
현재 널리 사용되고 있는 양자역학 원리를 이용하여 정확하면서도 계산시간이 획기적으로 개선된 새로운 전자밀도범함수 계산이론*이 국내 연구진에 의해 개발됐다.
*) 전자밀도범함수 계산이론 : 비교적 간단한 파동함수와 전자밀도만으로 에너지와 성질을 계산할 수 있음을 증명한 이론
우리 학교 EEWS대학원 정유성 교수(38세)와 윌리엄 고다드 교수가 주도한 이번 연구는 교육과학기술부(장관 이주호)와 한국연구재단이 추진하는 WCU(세계수준의 연구중심대학)육성사업의 지원을 받아 수행되었고, 연구결과는 자연과학분야의 권위 있는 학술지인 ‘미국립과학원회보(PNAS)’ 11월 23일자 온라인으로 게재되었다.
정유성 교수와 고다드 교수는 기존의 양자계산의 문제점인 계산시간과 부정확한 예측으로 인한 결과의 신뢰성이 떨어지는 단점을 보완하여, 정확하면서도 빠른 전자밀도범함수 이론과 알고리즘을 개발하였다.
양자역학 계산법 중 파동함수*를 이용하면 정확도가 높은 반면에 계산시간도 빠르게 증가해 수백-수천 개의 원자를 갖는 거대 분자에 적용하기 어렵고, 상대적으로 계산량이 적은 전자밀도를 변수로 사용할 경우 적용할 수 있는 분자의 크기는 증가하지만 정확도가 떨어지는 단점이 있었다.
*) 파동함수(波動函數) : 양자역학에서 물질입자인 전자·양성자·중성자 등의 상태를 나타내는 양
전자들의 상호작용은 스핀*이 같은 전자들끼리의 상호작용과 스핀이 다른 전자들끼리의 상호작용으로 나뉘는데, 파울리의 배타 원리에 의해 스핀이 다른 전자 사이의 거리가 더 가까우므로 스핀이 다른 전자들의 상호작용이 더 크다. 연구팀은 이 점에 착안하여, 기존에 존재하던 정확한 계산법에서 스핀이 다른 전자 사이의 상호작용을 중점적으로 계산하여 속도를 향상시켰다.
*) 스핀(spin) : 입자의 기본성질을 나타내는 물리량 중 하나로, 입자의 고유한 운동량을 나타냄. 소립자들의 특징을 밝히는 중요한 물리량임
또한 전자들의 상호작용은 국소성을 띠기 때문에 멀리 떨어진 전자 사이에는 상호작용이 거의 없어서 이들을 무시하더라도 총 에너지에 영향을 미치지 않는다는 점에 착안하여, 계산시간을 최대 100배이상 단축시키는 알고리즘을 개발하였다. 예를 들어, 기존의 계산방법으로는 탄소 200개와 수소 402개로 이루어진 알케인(aklane) 분자를 정확히 계산하는데 6개월이 걸린 반면, 새로운 방법론을 이용하면 비슷한 정확도로 하루(24시간)면 계산할 수 있다.
정유성 교수는 “그동안 국내의 계산과학 및 재료 설계 커뮤니티가 응용 연구에 주로 집중하여 짧은 시간 동안 훌륭한 결과를 많이 도출한 반면, 상대적으로 긴 시간을 요구하는 기초 방법론이나 소프트웨어 개발에서는 국제경쟁력이 뒤처져 있는 추세였다. 그러나 이번 연구는 기존의 방법들보다 월등한 정확도와 속도를 가진 방법론을 국내에서 개발했다는 점에서 의미가 크다”고 연구의의를 밝혔다.
아울러 이번에 개발된 방법론은 큐켐(Q-CHEM)이라는 상용 소프트웨어 패키지를 통해 일반연구자들에게 공급될 예정이다.알케인(alkane) 분자의 크기에 따라 본 연구에서 제시한 새로운 방법론(local XYGJ-OS)과 기존의 방법론의 계산 시간을 비교한 그래프. local XYGJ-OS는 전자 간 상호작용의 국소성을 이용해 계산 시간을 낮춘 방법이다.다양한 양자계산 방법과 본 연구에서 제시한 XYGJ-OS 방법의 오차. 233개 분자의 생성열을 실험값과 대조하여 오차의 절대값을 평균하였다. B2PLYP부터 XYG3까지의 방법 및 G2, G3 방법은 XYGJ-OS에 비하여 훨씬 많은 시간을 소모한다.
2011.11.29 조회수 17989