%EB%B0%A9%EB%B2%95%EB%A1%A0
-
 한재흥 교수팀, 미국기계학회 최우수논문상 수상
우리 대학 우주연구원장 항공우주공학과 한재흥 교수 연구팀이 미국기계학회(American Society of Mechanical Engineers, ASME)의 기계 디자인 저널(Journal of Mechanical Design)에서 2023년도 최우수 논문상(Best paper award)을 수상했다고 6일 밝혔다.
항공우주공학과 김태현 박사(주 저자)와 박사과정 장건익 학생, 이대영 교수(공동 저자)가 참여한 논문은 2023년도에 출판된 150여 편의 논문 중에서 기계설계(Machine design) 분야의 최고 우수 논문으로 선정됐으며, 국내 기관에서 수행한 연구 결과로 본 상을 받은 것은 이번이 최초다.
(제목: A Thickness-Accommodating Method for Void-Free Design in Uniformly Thick Origami)
연구진은 최근 항공우주 등 다양한 기계설계 분야에서 주목받고 있는 폴더블(Foldable) 구조 설계 시, 균일한 두께의 패널을 적용하면서도 구멍이나 빈 공간 없이 펼쳐진 상태의 유효 면적을 최대화할 수 있는 설계 방법론을 제시했다.
일반적인 구조는 재료가 두꺼워지면 간섭에 의해 구조물을 접는 것이 제한적이며, 조금만 패턴이 복잡해지더라도 빈 공간 없이 균일한 두께로 폴더블 구조를 설계하는 것은 어렵다고 알려져 있었다.
이러한 문제를 해결하고자 연구진은 균일한 두께의 재료가 적용 가능하면서도 빈 공간 없이 접을 수 있는 패턴 설계 방법론을 제시했다.
본 연구에서는 2차원 및 3차원의 패턴 설계 방법론을 체계적으로 제시했으며, 수학적인 분석을 통해 종이접기 패턴의 핵심적인 패턴 요소 중 하나인 D4V(Degree-4-Vertex)* 패턴에 대해 설계 방법론을 일반화했다. 이를 확장해 여러 D4V 요소로 구성된 구조를 두꺼운 패널로 제작하여 검증했다.
* D4V (Degree-4 Vertices): 네 개의 접힘선이 하나의 꼭짓점에서 모이는 패턴 기본 요소를 말하며, 다양한 형태의 오리가미 패턴에 적용되는 요소임
한재흥 교수는 “이번에 제시된 설계 방법론은 패널 두께에 제약받지 않으며 다양한 형태의 패턴에 적용할 수 있어 디스플레이와 같은 소형 구조부터 대형 전개형 건축물이나 우주 구조물 등에도 적용이 가능하다. 현재 이번 연구를 기반으로 달이나 화성 유인 탐사 시 사람과 장비를 보호하기 위한 미터급 전개형 우주 방호구조에 적용하여 개발하는 중이다”라고 말했다.
미국기계학회는 1880년도에 설립돼 130개 이상의 국가의 75,000명 이상의 정회원을 보유한 세계 최대의 국제 학술단체다. 현재 35개 이상의 학술 저널 및 학회 프로시딩을 출판하고 있으며, 전 세계에서 널리 사용되는 600개 이상의 표준을 제시하고 기계공학 분야를 주도하는 최고 권위의 학회다.
특히, 기계 디자인 저널은 1978년부터 출판돼, 자동화 설계, 메커니즘, 설계 방법론 등의 기계 시스템 설계 전반의 범위를 다루며 높은 학문적 영향력을 갖는 저널 중 하나다. 이 저널의 최고 우수 논문상은 매년 그 전 해 실린 모든 논문을 대상으로 편집위원회의 엄격한 평가 기준에 따라 오랜 심사를 거쳐 매년 1편에서 2편이 선정된다.
한편, 이번 연구는 과학기술정보통신부의 지원으로 한국연구재단의 지원을 받아 수행됐다.
2024.12.06 조회수 5427
한재흥 교수팀, 미국기계학회 최우수논문상 수상
우리 대학 우주연구원장 항공우주공학과 한재흥 교수 연구팀이 미국기계학회(American Society of Mechanical Engineers, ASME)의 기계 디자인 저널(Journal of Mechanical Design)에서 2023년도 최우수 논문상(Best paper award)을 수상했다고 6일 밝혔다.
항공우주공학과 김태현 박사(주 저자)와 박사과정 장건익 학생, 이대영 교수(공동 저자)가 참여한 논문은 2023년도에 출판된 150여 편의 논문 중에서 기계설계(Machine design) 분야의 최고 우수 논문으로 선정됐으며, 국내 기관에서 수행한 연구 결과로 본 상을 받은 것은 이번이 최초다.
(제목: A Thickness-Accommodating Method for Void-Free Design in Uniformly Thick Origami)
연구진은 최근 항공우주 등 다양한 기계설계 분야에서 주목받고 있는 폴더블(Foldable) 구조 설계 시, 균일한 두께의 패널을 적용하면서도 구멍이나 빈 공간 없이 펼쳐진 상태의 유효 면적을 최대화할 수 있는 설계 방법론을 제시했다.
일반적인 구조는 재료가 두꺼워지면 간섭에 의해 구조물을 접는 것이 제한적이며, 조금만 패턴이 복잡해지더라도 빈 공간 없이 균일한 두께로 폴더블 구조를 설계하는 것은 어렵다고 알려져 있었다.
이러한 문제를 해결하고자 연구진은 균일한 두께의 재료가 적용 가능하면서도 빈 공간 없이 접을 수 있는 패턴 설계 방법론을 제시했다.
본 연구에서는 2차원 및 3차원의 패턴 설계 방법론을 체계적으로 제시했으며, 수학적인 분석을 통해 종이접기 패턴의 핵심적인 패턴 요소 중 하나인 D4V(Degree-4-Vertex)* 패턴에 대해 설계 방법론을 일반화했다. 이를 확장해 여러 D4V 요소로 구성된 구조를 두꺼운 패널로 제작하여 검증했다.
* D4V (Degree-4 Vertices): 네 개의 접힘선이 하나의 꼭짓점에서 모이는 패턴 기본 요소를 말하며, 다양한 형태의 오리가미 패턴에 적용되는 요소임
한재흥 교수는 “이번에 제시된 설계 방법론은 패널 두께에 제약받지 않으며 다양한 형태의 패턴에 적용할 수 있어 디스플레이와 같은 소형 구조부터 대형 전개형 건축물이나 우주 구조물 등에도 적용이 가능하다. 현재 이번 연구를 기반으로 달이나 화성 유인 탐사 시 사람과 장비를 보호하기 위한 미터급 전개형 우주 방호구조에 적용하여 개발하는 중이다”라고 말했다.
미국기계학회는 1880년도에 설립돼 130개 이상의 국가의 75,000명 이상의 정회원을 보유한 세계 최대의 국제 학술단체다. 현재 35개 이상의 학술 저널 및 학회 프로시딩을 출판하고 있으며, 전 세계에서 널리 사용되는 600개 이상의 표준을 제시하고 기계공학 분야를 주도하는 최고 권위의 학회다.
특히, 기계 디자인 저널은 1978년부터 출판돼, 자동화 설계, 메커니즘, 설계 방법론 등의 기계 시스템 설계 전반의 범위를 다루며 높은 학문적 영향력을 갖는 저널 중 하나다. 이 저널의 최고 우수 논문상은 매년 그 전 해 실린 모든 논문을 대상으로 편집위원회의 엄격한 평가 기준에 따라 오랜 심사를 거쳐 매년 1편에서 2편이 선정된다.
한편, 이번 연구는 과학기술정보통신부의 지원으로 한국연구재단의 지원을 받아 수행됐다.
2024.12.06 조회수 5427 -
 암 유발 물질 컴퓨터로 예측하다
암은 정상세포와 다르게 세포 내 비정상적인 축적을 통해 유발되는 대사 반응을 하며, 암의 치료 및 진단을 목적으로 이런 암 대사반응에 대해 다방면으로 연구되고 있다. 이에 우리 대학 연구진이 컴퓨터를 통해 24개 암종에 해당하는 1,043명의 암 환자에 대한 대사 모델 구축에 성공했다.
우리 대학 생명화학공학과 김현욱 교수, 이상엽 특훈교수 연구팀이 서울대학교병원 고영일 교수, 윤홍석 교수 및 정창욱 교수 연구팀과의 공동연구를 통해, 암 체세포 유전자 돌연변이와 연관된 새로운 대사물질 및 대사경로를 예측하는 컴퓨터 방법론을 개발했다고 18일 밝혔다.
최근 암 유발 대사물질(oncometabolite)*의 발견과 이를 표적으로 하는 신약들이 미국식품의약국(FDA)의 승인을 받으며 주목받고 있는데, 이에는 급성 골수성 백혈병의 치료제로 사용되고 있는 ‘팁소보(성분명: 아이보시데닙)’ 및 약물 ‘아이드하이파(성분명: 에나시데닙)’가 포함된다.
*암 유발 대사물질 (oncometabolite): 세포 내 비정상적인 축적을 통해 암을 유발하는 대사물질. 이러한 대사물질들은 특정 유전자 돌연변이의 영향으로 대사 과정 중에 비정상적으로 높은 농도로 축적되며, 이러한 축적은 암세포의 성장과 생존을 촉진함. 기존 연구에서 확인된 주요 암 유발 대사물질로는 2-하이드록시글루타레이트(2-hydroxyglutarate), 숙시네이트(succinate), 푸마레이트(fumarate) 등이 보고됨.
하지만, 암 대사 연구와 새로운 암 유발 대사물질 발굴에는 대사체학 등의 방법론이 필요하며, 이를 대규모 환자 샘플에 적용하기 위해서는 상당한 시간과 비용이 소요된다. 이러한 이유로, 암과 관련된 많은 유전자 돌연변이들이 밝혀졌음에도, 그에 상응하는 암 유발 대사물질은 극소수만 알려져 있다.
김현욱 교수 공동연구팀은 세포 대사 정보를 예측할 수 있는 게놈 수준의 대사 모델*에 국제 암 연구 컨소시엄에서 공개하고 있는 암 환자들의 전사체 데이터를 통합해, 24개 암종에 해당하는 1,043명의 암 환자에 대한 대사 모델을 성공적으로 구축했다.
*게놈 수준의 대사모델: 세포의 전체 대사 네트워크를 다루는 컴퓨터 모델로서, 세포 내 모든 대사반응에 대한 정보가 담겨 있으며, 다양한 조건에서 세포의 대사 활성을 예측하는 것이 가능
공동연구팀은 1,043명의 암 환자 특이 대사 모델과 동일 환자들의 암 체세포 돌연변이 데이터를 활용해, 다음의 4단계로 구성된 컴퓨터 방법론을 개발했다 (그림 1). 첫 단계에서는 암 환자 특이 대사 모델을 시뮬레이션해, 환자 별로 모든 대사물질들의 활성을 예측한다. 두 번째 단계로는 특정 유전자 돌연변이가 앞서 예측된 대사물질의 활성에 유의한 차이를 일으키는 짝을 선별한다. 세 번째 단계로, 특정 유전자 돌연변이와 연결된 대사물질들을 대상으로, 이들과 유의하게 연관된 대사경로를 추가로 선별한다. 마지막 단계로서, ‘유전자-대사물질-대사경로’ 조합을 완성해, 컴퓨터 방법론 결과로써 도출하게 된다.
이번 논문의 공동 제1 저자인 이가령 박사(現 다나파버 암센터 및 하버드 의과대학 박사후연구원)와 이상미 박사(現 하버드 의과대학 박사후연구원)는 “이번 연구에서 개발된 방법론은 암 환자 코호트의 돌연변이 및 전사체 데이터를 토대로 다른 암종에 대해서도 쉽게 적용될 수 있으며, 유전자 돌연변이가 대사경로를 통해 어떻게 세포대사에 변화를 일으키는지 체계적으로 예측할 수 있는 최초의 컴퓨터 방법론이라는 데 큰 의의가 있다” 한다고 말했다.
또한 김현욱 교수는 “이번 공동연구의 결과는 향후 암 대사 및 암 유발 대사물질 연구에서 중요한 참고 자료로 활용될 수 있을 것”이라고 강조했다.
한편 이번 논문은 바이오메드 센트럴(BioMed Central) 社가 발행하며, 생명공학 및 유전학 분야의 대표적 국제학술지인 게놈 바이올로지(Genome Biology, JCR 분야 상위 5% 이내)에 게재됐다.
※ 논문명 : Prediction of metabolites associated with somatic mutations in cancers by using genome-scale metabolic models and mutation data
※ 저자 정보 : 이가령(한국과학기술원, 공동 제1 저자), 이상미(한국과학기술원, 공동 제1 저자), 이성영(서울대학교병원, 공동저자), 정창욱(서울대학교병원, 공동저자), 송효진(서울대학교병원, 공동저자), 이상엽(한국과학기술원, 공동저자), 윤홍석(서울대학교병원, 교신저자), 고영일(서울대학교병원, 교신저자), 김현욱(한국과학기술원, 교신저자) 포함 총 9명
이번 연구는 과학기술정보통신부 한국연구재단의 지원을 받아 수행됐다.
2024.03.18 조회수 7606
암 유발 물질 컴퓨터로 예측하다
암은 정상세포와 다르게 세포 내 비정상적인 축적을 통해 유발되는 대사 반응을 하며, 암의 치료 및 진단을 목적으로 이런 암 대사반응에 대해 다방면으로 연구되고 있다. 이에 우리 대학 연구진이 컴퓨터를 통해 24개 암종에 해당하는 1,043명의 암 환자에 대한 대사 모델 구축에 성공했다.
우리 대학 생명화학공학과 김현욱 교수, 이상엽 특훈교수 연구팀이 서울대학교병원 고영일 교수, 윤홍석 교수 및 정창욱 교수 연구팀과의 공동연구를 통해, 암 체세포 유전자 돌연변이와 연관된 새로운 대사물질 및 대사경로를 예측하는 컴퓨터 방법론을 개발했다고 18일 밝혔다.
최근 암 유발 대사물질(oncometabolite)*의 발견과 이를 표적으로 하는 신약들이 미국식품의약국(FDA)의 승인을 받으며 주목받고 있는데, 이에는 급성 골수성 백혈병의 치료제로 사용되고 있는 ‘팁소보(성분명: 아이보시데닙)’ 및 약물 ‘아이드하이파(성분명: 에나시데닙)’가 포함된다.
*암 유발 대사물질 (oncometabolite): 세포 내 비정상적인 축적을 통해 암을 유발하는 대사물질. 이러한 대사물질들은 특정 유전자 돌연변이의 영향으로 대사 과정 중에 비정상적으로 높은 농도로 축적되며, 이러한 축적은 암세포의 성장과 생존을 촉진함. 기존 연구에서 확인된 주요 암 유발 대사물질로는 2-하이드록시글루타레이트(2-hydroxyglutarate), 숙시네이트(succinate), 푸마레이트(fumarate) 등이 보고됨.
하지만, 암 대사 연구와 새로운 암 유발 대사물질 발굴에는 대사체학 등의 방법론이 필요하며, 이를 대규모 환자 샘플에 적용하기 위해서는 상당한 시간과 비용이 소요된다. 이러한 이유로, 암과 관련된 많은 유전자 돌연변이들이 밝혀졌음에도, 그에 상응하는 암 유발 대사물질은 극소수만 알려져 있다.
김현욱 교수 공동연구팀은 세포 대사 정보를 예측할 수 있는 게놈 수준의 대사 모델*에 국제 암 연구 컨소시엄에서 공개하고 있는 암 환자들의 전사체 데이터를 통합해, 24개 암종에 해당하는 1,043명의 암 환자에 대한 대사 모델을 성공적으로 구축했다.
*게놈 수준의 대사모델: 세포의 전체 대사 네트워크를 다루는 컴퓨터 모델로서, 세포 내 모든 대사반응에 대한 정보가 담겨 있으며, 다양한 조건에서 세포의 대사 활성을 예측하는 것이 가능
공동연구팀은 1,043명의 암 환자 특이 대사 모델과 동일 환자들의 암 체세포 돌연변이 데이터를 활용해, 다음의 4단계로 구성된 컴퓨터 방법론을 개발했다 (그림 1). 첫 단계에서는 암 환자 특이 대사 모델을 시뮬레이션해, 환자 별로 모든 대사물질들의 활성을 예측한다. 두 번째 단계로는 특정 유전자 돌연변이가 앞서 예측된 대사물질의 활성에 유의한 차이를 일으키는 짝을 선별한다. 세 번째 단계로, 특정 유전자 돌연변이와 연결된 대사물질들을 대상으로, 이들과 유의하게 연관된 대사경로를 추가로 선별한다. 마지막 단계로서, ‘유전자-대사물질-대사경로’ 조합을 완성해, 컴퓨터 방법론 결과로써 도출하게 된다.
이번 논문의 공동 제1 저자인 이가령 박사(現 다나파버 암센터 및 하버드 의과대학 박사후연구원)와 이상미 박사(現 하버드 의과대학 박사후연구원)는 “이번 연구에서 개발된 방법론은 암 환자 코호트의 돌연변이 및 전사체 데이터를 토대로 다른 암종에 대해서도 쉽게 적용될 수 있으며, 유전자 돌연변이가 대사경로를 통해 어떻게 세포대사에 변화를 일으키는지 체계적으로 예측할 수 있는 최초의 컴퓨터 방법론이라는 데 큰 의의가 있다” 한다고 말했다.
또한 김현욱 교수는 “이번 공동연구의 결과는 향후 암 대사 및 암 유발 대사물질 연구에서 중요한 참고 자료로 활용될 수 있을 것”이라고 강조했다.
한편 이번 논문은 바이오메드 센트럴(BioMed Central) 社가 발행하며, 생명공학 및 유전학 분야의 대표적 국제학술지인 게놈 바이올로지(Genome Biology, JCR 분야 상위 5% 이내)에 게재됐다.
※ 논문명 : Prediction of metabolites associated with somatic mutations in cancers by using genome-scale metabolic models and mutation data
※ 저자 정보 : 이가령(한국과학기술원, 공동 제1 저자), 이상미(한국과학기술원, 공동 제1 저자), 이성영(서울대학교병원, 공동저자), 정창욱(서울대학교병원, 공동저자), 송효진(서울대학교병원, 공동저자), 이상엽(한국과학기술원, 공동저자), 윤홍석(서울대학교병원, 교신저자), 고영일(서울대학교병원, 교신저자), 김현욱(한국과학기술원, 교신저자) 포함 총 9명
이번 연구는 과학기술정보통신부 한국연구재단의 지원을 받아 수행됐다.
2024.03.18 조회수 7606 -
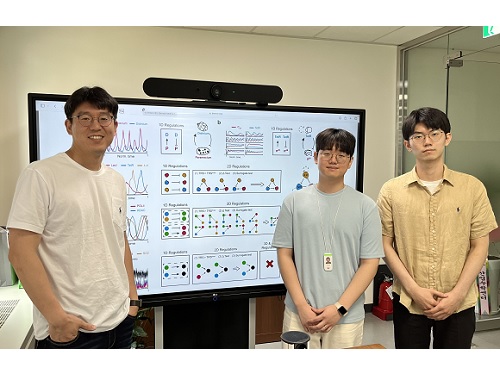 인과관계 추정 정확도 높인 새로운 방법론 개발
우리 대학 수리과학과 김재경 교수 연구팀이 수학 모델을 기반으로 시계열 데이터의 인과관계를 추정하는 새로운 방법론을 개발했다. 복잡한 계산 과정을 없애 기존보다 빠른 속도로 추론이 가능하면서도, 정확도는 획기적으로 높였다.
매 순간 다양한 데이터가 기록되고 있다. 그중 시간의 흐름을 기준으로 기록된 ‘시계열 데이터’는 일기 예보와 경제 분야뿐만 아니라 의학 분야에서도 가치 있게 쓰인다. 입원 환자의 심전도 측정을 통해 심장 발작의 직접적인 요인을 찾는 것과 같이 인과관계를 추정하는 것이 대표적이다. 최근에는 스마트 워치 등 웨어러블 기기를 통해 일상에서 건강 데이터를 쉽게 수집할 수 있게 되면서, 의학 분야에서 시계열 데이터 분석의 중요성이 더 커지고 있다.
시계열 데이터에서 인과관계를 추정하는 대표적인 방법으로는 2003년 노벨 경제학상을 수상한 클라이브 그레인저 미국 샌디에이고캘리포니아대(UC샌디에이고) 교수가 제시한 ‘그레인저 인과관계 검정(Granger causality test)’이 있다. 이는 미래 경제지표 예측, 질병 요인분석, 지구온난화의 원인 등 수많은 분야에 걸쳐 응용됐다. 그레인저 인과관계 검정을 개선한 정보 이론 기반의 다양한 인과관계 추정 방법이 개발됐지만, 일련의 방법들은 시계열 데이터가 비슷한 주기로 변화하는 동시성을 가지기만 하면, 인과관계가 있다고 잘못 예측하는 경우가 많았다. 또한, 직접적인 인과관계와 간접적인 인과관계를 구별하지 못한다는 한계도 있었다.
이러한 한계를 극복하기 위해 최근 수리 모델을 기반으로 하는 방법론들이 등장했다. 수리 모델로 주어진 시계열 데이터를 잘 맞출 수 있는지 확인하는 방법을 통해 인과관계를 예측한다. 수리 모델이 정확하기만 하면 기존 그레인저 인과관계 검정의 한계인 동시성과 간접적인 영향을 인과관계와 혼동하지 않는다는 장점이 있다. 그러나 정확한 수리 모델을 알기 힘들고, 현재까지 제시된 수리 모델 기반 방법론들은 복잡한 계산이 필요해 추정 시간이 많이 걸린다는 단점이 있다.
이러한 상황에서 연구팀은 기존 방법론들의 한계를 모두 해결한 새로운 방법론 ‘GOBI(General ODE-Based Inference)’를 개발했다. 우선, 연구팀은 시계열 데이터가 일반적인 수학 모델로 표현될 수 있는지 확인하는 수학 이론을 만들었다. 그리고 이 이론을 바탕으로 정확한 수리 모델이나 복잡한 계산 없이도 시계열 데이터로부터 인과관계를 추정하는 방법론을 개발했다.
개발한 방법론을 인과관계 분석에 적용해 본 결과 세포 내 분자들의 상호작용, 생태계 네트워크, 기상 시스템 등 다양한 분야의 데이터에서 기존 방법론에 비해 월등한 성능을 보여줬다. 특히, 동시성 및 간접적인 영향을 가지는 시계열 데이터에서도 인과관계를 성공적으로 추론했다. 연구진은 GOBI를 통해서 여러 오염 물질 중 이산화질소와 호흡기로 유입되는 부유 미립자(직경 10㎛ 이하의 입자)가 심혈관계 질환에 영향을 미친다는 것을 확인할 수 있었다.
김재경 교수는 “수학과 통계를 결합하여 정확하면서도 다양한 시스템에 유연하게 적용할 수 있는 새로운 인과관계 추정 방법론을 개발했다”며 “사회 및 자연과학 분야에 걸쳐 두루 사용되는 인과관계 추정 연구에 새로운 패러다임을 제시할 것으로 예상된다”고 말했다.
연구결과는 7월 24일 국제학술지 ‘네이처 커뮤니케이션즈(Nature Communications, IF 17.694)’ 온라인판에 실렸으며, 우리 대학 박세호 학사과정(제1저자)과 하석민 학사과정(제2저자)이 참여했다.
2023.07.26 조회수 9213
인과관계 추정 정확도 높인 새로운 방법론 개발
우리 대학 수리과학과 김재경 교수 연구팀이 수학 모델을 기반으로 시계열 데이터의 인과관계를 추정하는 새로운 방법론을 개발했다. 복잡한 계산 과정을 없애 기존보다 빠른 속도로 추론이 가능하면서도, 정확도는 획기적으로 높였다.
매 순간 다양한 데이터가 기록되고 있다. 그중 시간의 흐름을 기준으로 기록된 ‘시계열 데이터’는 일기 예보와 경제 분야뿐만 아니라 의학 분야에서도 가치 있게 쓰인다. 입원 환자의 심전도 측정을 통해 심장 발작의 직접적인 요인을 찾는 것과 같이 인과관계를 추정하는 것이 대표적이다. 최근에는 스마트 워치 등 웨어러블 기기를 통해 일상에서 건강 데이터를 쉽게 수집할 수 있게 되면서, 의학 분야에서 시계열 데이터 분석의 중요성이 더 커지고 있다.
시계열 데이터에서 인과관계를 추정하는 대표적인 방법으로는 2003년 노벨 경제학상을 수상한 클라이브 그레인저 미국 샌디에이고캘리포니아대(UC샌디에이고) 교수가 제시한 ‘그레인저 인과관계 검정(Granger causality test)’이 있다. 이는 미래 경제지표 예측, 질병 요인분석, 지구온난화의 원인 등 수많은 분야에 걸쳐 응용됐다. 그레인저 인과관계 검정을 개선한 정보 이론 기반의 다양한 인과관계 추정 방법이 개발됐지만, 일련의 방법들은 시계열 데이터가 비슷한 주기로 변화하는 동시성을 가지기만 하면, 인과관계가 있다고 잘못 예측하는 경우가 많았다. 또한, 직접적인 인과관계와 간접적인 인과관계를 구별하지 못한다는 한계도 있었다.
이러한 한계를 극복하기 위해 최근 수리 모델을 기반으로 하는 방법론들이 등장했다. 수리 모델로 주어진 시계열 데이터를 잘 맞출 수 있는지 확인하는 방법을 통해 인과관계를 예측한다. 수리 모델이 정확하기만 하면 기존 그레인저 인과관계 검정의 한계인 동시성과 간접적인 영향을 인과관계와 혼동하지 않는다는 장점이 있다. 그러나 정확한 수리 모델을 알기 힘들고, 현재까지 제시된 수리 모델 기반 방법론들은 복잡한 계산이 필요해 추정 시간이 많이 걸린다는 단점이 있다.
이러한 상황에서 연구팀은 기존 방법론들의 한계를 모두 해결한 새로운 방법론 ‘GOBI(General ODE-Based Inference)’를 개발했다. 우선, 연구팀은 시계열 데이터가 일반적인 수학 모델로 표현될 수 있는지 확인하는 수학 이론을 만들었다. 그리고 이 이론을 바탕으로 정확한 수리 모델이나 복잡한 계산 없이도 시계열 데이터로부터 인과관계를 추정하는 방법론을 개발했다.
개발한 방법론을 인과관계 분석에 적용해 본 결과 세포 내 분자들의 상호작용, 생태계 네트워크, 기상 시스템 등 다양한 분야의 데이터에서 기존 방법론에 비해 월등한 성능을 보여줬다. 특히, 동시성 및 간접적인 영향을 가지는 시계열 데이터에서도 인과관계를 성공적으로 추론했다. 연구진은 GOBI를 통해서 여러 오염 물질 중 이산화질소와 호흡기로 유입되는 부유 미립자(직경 10㎛ 이하의 입자)가 심혈관계 질환에 영향을 미친다는 것을 확인할 수 있었다.
김재경 교수는 “수학과 통계를 결합하여 정확하면서도 다양한 시스템에 유연하게 적용할 수 있는 새로운 인과관계 추정 방법론을 개발했다”며 “사회 및 자연과학 분야에 걸쳐 두루 사용되는 인과관계 추정 연구에 새로운 패러다임을 제시할 것으로 예상된다”고 말했다.
연구결과는 7월 24일 국제학술지 ‘네이처 커뮤니케이션즈(Nature Communications, IF 17.694)’ 온라인판에 실렸으며, 우리 대학 박세호 학사과정(제1저자)과 하석민 학사과정(제2저자)이 참여했다.
2023.07.26 조회수 9213 -
 서양 미술사 빅데이터 분석으로 회화 속 구도 변화 규명
우리 대학 물리학과 정하웅 교수 연구팀이 충북대학교 물리학과 한승기 교수 연구팀과 공동연구를 통해 르네상스부터 동시대 미술에 이르기까지 약 500년에 걸친 풍경화 1만 5천여 점을 정보이론과 네트워크 이론으로 분석해 서양 미술사 속 풍경화의 구도와 구성 비율의 점진적 변화를 수치적으로 규명했다.
우리 대학 물리학과 이병휘 박사과정 학생과 충북대 서민경 학생이 주도한 이번 연구는 세계적인 학술지 ‘미국 국립과학원회보(Proceedings of the National Academy of Sciences of the USA, 이하 PNAS)’에 10월 117권 43호에 출판됬다. (논문명: Dissecting Landscape Art History with Information Theory, 정보이론으로 해부한 풍경화의 역사). 해당 논문은 PNAS의 In this issue 섹션에 이번 호의 대표 논문으로 선정되었고, 코멘터리와 함께 게재됐다.
화가는 그림을 그릴 때 선, 색, 형태, 모양 등 여러 가지 시각적 구성 요소들을 다양한 ‘구성 원리’를 바탕으로 조화로운 최종 작품을 완성한다. 미술사와 미학 연구자들은 작가들이 작품을 생성할 때 잠재적으로 적용한 구성 원리가 시대와 문화를 초월하는 공통적인 특징을 가지는지, 혹은 시대나 문화적 환경에 따라 어떻게 달라지는지 이해하고자 시도해왔다. 특별히 대표적인 구성 원리중 하나인 작품구도 속 사용된 ‘비례’와 ‘비율’은 미술사가들과 미학자들의 오랜 관심사였다. 역사적으로 많은 논란을 일으킨 사례로는 황금비(Golden ratio)가 있다. 기원전 300년 전 유클리드의 원론에 의해 처음 제시된 황금비는 1500년대 초 이탈리아의 수학자 루카 파치올리의 책을 통해 ‘신성한 비율’이라는 이름으로 대중적으로 소개되며 유명해졌다. 최근까지도 황금비의 미적 선호도에 관한 논란은 계속되어 왔는데, 파르테논 신전이나 밀로의 비너스 등 여러 아름다운 미술 작품 속에 황금비가 발견되었다는 대부분의 주장들은 오늘날 근거가 부족한 것으로 밝혀지고 있다. 그렇다면 미술사 속에서 화가들이 특별히 선호한 비율은 과연 존재했을까? 혹은 시대에 따라 선호한 비율은 어떻게 변해왔을까?
연구팀은 회화 속 색상의 공간적 배치를 특징짓는 정보이론적 분할 방법론을 적용해 서양 미술사 풍경화 역사 속에서 사용된 구도와 구성 비율을 수치화하는 방법을 제시했다. (그림1 참조) *두 가지 대규모 온라인 갤러리 로부터 16세기 르네상스 시대부터 20세기 미술까지 500년 이상의 시간에 걸친 서양 미술사 속 풍경화 1만 5천여 점을 수집하여 분석한 결과, 화가들이 선호한 거시적 작품 구도와 구성 비율이 시대에 따라 일정하거나 무작위적이지 않고, 점진적이고 체계적인 변화과정을 거쳐왔음을 확인했다.
* 온라인 시각 예술 백과사전인 위키 아트(‘WikiArt’)와 헝가리 부다베스트 물리학 컴퓨터 네트워킹 연구센터에서 운영하는 온라인 갤러리인 웹 갤러리 오브 아트(‘Web Gallery of Art’)의 풍경화 데이터를 활용
연구팀은 먼저 정보이론적 분할 방법론을 이용해 풍경화 구도를 특징지었는데, 16세기부터 19세기 중반까지의 풍경화는 지배적인 수평 구조와 수직 구조가 함께 존재하는 ‘수평-수직’ 형태의 구도가 가장 빈번하게 사용되었으나, 시간이 흐를수록 전경-중경-후경과 같이 두 개의 수평 구조가 존재하는 ‘수평-수평’ 형태의 구도 사용이 점차 증가해 19세기 중반 이후부터는 ‘수평-수평’ 형태의 구도가 가장 지배적인 구도가 되었음을 확인했다. (그림 2 참조) 흥미롭게도 이러한 시간에 따른 구도 변화 패턴은 여러 국적에 걸쳐서도 유사하게 나타났다.
또한 연구팀은 색상 사용 패턴이 급격하게 달라지는 지배적인 수평선의 위치를 기반으로 시대와 작가별로 풍경 구도를 잡는데 자주 사용한 구성 비율을 측정했는데, 선호된 구성 비율은 시간에 따라 매우 점진적이고, 부드러운 변화 과정을 보였다. 작가들의 선호한 풍경화 속 지배적인 수평선은 바로크 시대 17세기 무렵 그림의 절반 아래에 해당하는 낮은 위치에서 발견되었으나, 그 후 점차 위쪽으로 움직여 19세기 이후에는 작품 위에서부터 1/3 지점에서 가장 많은 빈도로 발견됐다. 신기하게도 1/3 구성 비율을 가장 빈번하게 사용하는 특징은 다양한 현대 미술 주의(ism)에 걸쳐 유사하게 발견됐는데, 이러한 발견은 미술 양식의 폭발적인 다양성을 대표하는 현대 미술의 여러 주의들이 색채 사용과 표현 방법에선 다양성과 차별성을 추구했으나, 구도와 구성 비율의 관점에서는 유사한 사용 패턴을 보였다는 점에서 새로운 발견이다.
연구팀은 또한 네트워크 과학 방법론을 적용해 서로 유사한 구도를 적용한 작가들과 사조들로 이루어진 네트워크를 구축하여 분석했다. 이 작가-사조 네트워크는 크게 세 가지 거대 군집으로 구성돼 있었는데, 신기하게도 구도 사용의 유사성만을 바탕으로 한 작가들과 사조 속 군집은 시기적으로도 근접한 시기에 활동을 보인 작가들과 사조들로 이루어져 있었다. 이는 기존 알려진 개별 작가들의 생애와 개별 사조의 시간 범위를 초월하는 미술사 구도 양식 속 거대 군집이 있음을 시사한다.
정하웅 교수는 ‘이 같이 시대에 따른 깔끔하고 체계적인 서양 미술사 속 구도변화는 미술의 실제 역사의 모습을 반영하고 있을 수도 있지만, 동시에 높을 확률로 그동안 미술사가들과 비평가들에 의해 평가되고 정리돼 온 주류 미술사의 편향을 나타내고 있을 수 있음을 주의해야 한다’고 지적했다.
한편 이번 연구는 한국연구재단의 지원을 통해 수행됐다.
2020.11.02 조회수 33664
서양 미술사 빅데이터 분석으로 회화 속 구도 변화 규명
우리 대학 물리학과 정하웅 교수 연구팀이 충북대학교 물리학과 한승기 교수 연구팀과 공동연구를 통해 르네상스부터 동시대 미술에 이르기까지 약 500년에 걸친 풍경화 1만 5천여 점을 정보이론과 네트워크 이론으로 분석해 서양 미술사 속 풍경화의 구도와 구성 비율의 점진적 변화를 수치적으로 규명했다.
우리 대학 물리학과 이병휘 박사과정 학생과 충북대 서민경 학생이 주도한 이번 연구는 세계적인 학술지 ‘미국 국립과학원회보(Proceedings of the National Academy of Sciences of the USA, 이하 PNAS)’에 10월 117권 43호에 출판됬다. (논문명: Dissecting Landscape Art History with Information Theory, 정보이론으로 해부한 풍경화의 역사). 해당 논문은 PNAS의 In this issue 섹션에 이번 호의 대표 논문으로 선정되었고, 코멘터리와 함께 게재됐다.
화가는 그림을 그릴 때 선, 색, 형태, 모양 등 여러 가지 시각적 구성 요소들을 다양한 ‘구성 원리’를 바탕으로 조화로운 최종 작품을 완성한다. 미술사와 미학 연구자들은 작가들이 작품을 생성할 때 잠재적으로 적용한 구성 원리가 시대와 문화를 초월하는 공통적인 특징을 가지는지, 혹은 시대나 문화적 환경에 따라 어떻게 달라지는지 이해하고자 시도해왔다. 특별히 대표적인 구성 원리중 하나인 작품구도 속 사용된 ‘비례’와 ‘비율’은 미술사가들과 미학자들의 오랜 관심사였다. 역사적으로 많은 논란을 일으킨 사례로는 황금비(Golden ratio)가 있다. 기원전 300년 전 유클리드의 원론에 의해 처음 제시된 황금비는 1500년대 초 이탈리아의 수학자 루카 파치올리의 책을 통해 ‘신성한 비율’이라는 이름으로 대중적으로 소개되며 유명해졌다. 최근까지도 황금비의 미적 선호도에 관한 논란은 계속되어 왔는데, 파르테논 신전이나 밀로의 비너스 등 여러 아름다운 미술 작품 속에 황금비가 발견되었다는 대부분의 주장들은 오늘날 근거가 부족한 것으로 밝혀지고 있다. 그렇다면 미술사 속에서 화가들이 특별히 선호한 비율은 과연 존재했을까? 혹은 시대에 따라 선호한 비율은 어떻게 변해왔을까?
연구팀은 회화 속 색상의 공간적 배치를 특징짓는 정보이론적 분할 방법론을 적용해 서양 미술사 풍경화 역사 속에서 사용된 구도와 구성 비율을 수치화하는 방법을 제시했다. (그림1 참조) *두 가지 대규모 온라인 갤러리 로부터 16세기 르네상스 시대부터 20세기 미술까지 500년 이상의 시간에 걸친 서양 미술사 속 풍경화 1만 5천여 점을 수집하여 분석한 결과, 화가들이 선호한 거시적 작품 구도와 구성 비율이 시대에 따라 일정하거나 무작위적이지 않고, 점진적이고 체계적인 변화과정을 거쳐왔음을 확인했다.
* 온라인 시각 예술 백과사전인 위키 아트(‘WikiArt’)와 헝가리 부다베스트 물리학 컴퓨터 네트워킹 연구센터에서 운영하는 온라인 갤러리인 웹 갤러리 오브 아트(‘Web Gallery of Art’)의 풍경화 데이터를 활용
연구팀은 먼저 정보이론적 분할 방법론을 이용해 풍경화 구도를 특징지었는데, 16세기부터 19세기 중반까지의 풍경화는 지배적인 수평 구조와 수직 구조가 함께 존재하는 ‘수평-수직’ 형태의 구도가 가장 빈번하게 사용되었으나, 시간이 흐를수록 전경-중경-후경과 같이 두 개의 수평 구조가 존재하는 ‘수평-수평’ 형태의 구도 사용이 점차 증가해 19세기 중반 이후부터는 ‘수평-수평’ 형태의 구도가 가장 지배적인 구도가 되었음을 확인했다. (그림 2 참조) 흥미롭게도 이러한 시간에 따른 구도 변화 패턴은 여러 국적에 걸쳐서도 유사하게 나타났다.
또한 연구팀은 색상 사용 패턴이 급격하게 달라지는 지배적인 수평선의 위치를 기반으로 시대와 작가별로 풍경 구도를 잡는데 자주 사용한 구성 비율을 측정했는데, 선호된 구성 비율은 시간에 따라 매우 점진적이고, 부드러운 변화 과정을 보였다. 작가들의 선호한 풍경화 속 지배적인 수평선은 바로크 시대 17세기 무렵 그림의 절반 아래에 해당하는 낮은 위치에서 발견되었으나, 그 후 점차 위쪽으로 움직여 19세기 이후에는 작품 위에서부터 1/3 지점에서 가장 많은 빈도로 발견됐다. 신기하게도 1/3 구성 비율을 가장 빈번하게 사용하는 특징은 다양한 현대 미술 주의(ism)에 걸쳐 유사하게 발견됐는데, 이러한 발견은 미술 양식의 폭발적인 다양성을 대표하는 현대 미술의 여러 주의들이 색채 사용과 표현 방법에선 다양성과 차별성을 추구했으나, 구도와 구성 비율의 관점에서는 유사한 사용 패턴을 보였다는 점에서 새로운 발견이다.
연구팀은 또한 네트워크 과학 방법론을 적용해 서로 유사한 구도를 적용한 작가들과 사조들로 이루어진 네트워크를 구축하여 분석했다. 이 작가-사조 네트워크는 크게 세 가지 거대 군집으로 구성돼 있었는데, 신기하게도 구도 사용의 유사성만을 바탕으로 한 작가들과 사조 속 군집은 시기적으로도 근접한 시기에 활동을 보인 작가들과 사조들로 이루어져 있었다. 이는 기존 알려진 개별 작가들의 생애와 개별 사조의 시간 범위를 초월하는 미술사 구도 양식 속 거대 군집이 있음을 시사한다.
정하웅 교수는 ‘이 같이 시대에 따른 깔끔하고 체계적인 서양 미술사 속 구도변화는 미술의 실제 역사의 모습을 반영하고 있을 수도 있지만, 동시에 높을 확률로 그동안 미술사가들과 비평가들에 의해 평가되고 정리돼 온 주류 미술사의 편향을 나타내고 있을 수 있음을 주의해야 한다’고 지적했다.
한편 이번 연구는 한국연구재단의 지원을 통해 수행됐다.
2020.11.02 조회수 33664 -
 이상엽 특훈교수, 김현욱 교수, 인공지능 이용한 효소기능 예측 기술 개발
우리 대학 생명화학공학과 이상엽 특훈교수와 김현욱 교수의 초세대 협업연구실 공동연구팀이 딥러닝(deep learning) 기술을 이용해 효소의 기능을 신속하고 정확하게 예측할 수 있는 컴퓨터 방법론 DeepEC를 개발했다.
공동연구팀의 류재용 박사가 1 저자로 참여한 이번 연구결과는 국제학술지 ‘미국 국립과학원 회보(PNAS)’ 6월 20일 자 온라인판에 게재됐다. (논문명 : Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers)
효소는 세포 내의 생화학반응들을 촉진하는 단백질 촉매로 이들의 기능을 정확히 이해하는 것은 세포의 대사(metabolism) 과정을 이해하는 데에 매우 중요하다.
특히 효소들은 다양한 질병 발생 원리 및 산업 생명공학과 밀접한 연관이 있어 방대한 게놈 정보에서 효소들의 기능을 빠르고 정확하게 예측하는 기술은 응용기술 측면에서도 중요하다.
효소의 기능을 표기하는 시스템 중 대표적인 것이 EC 번호(enzyme commission number)이다. EC 번호는 ‘EC 3.4.11.4’처럼 효소가 매개하는 생화학반응들의 종류에 따라 총 4개의 숫자로 구성돼 있다.
중요한 것은 특정 효소에 주어진 EC 번호를 통해서 해당 효소가 어떠한 종류의 생화학반응을 매개하는지 알 수 있다는 것이다. 따라서 게놈으로부터 얻을 수 있는 효소 단백질 서열의 EC 번호를 빠르고 정확하게 예측할 수 있는 기술은 효소 및 대사 관련 문제를 해결하는 데 중요한 역할을 한다.
작년까지 여러 해에 걸쳐 EC 번호를 예측해주는 컴퓨터 방법론들이 최소 10개 이상 개발됐다. 그러나 이들 모두 예측 속도, 예측 정확성 및 예측 가능 범위 측면에서 발전 필요성이 있었다. 특히 현대 생명과학 및 생명공학에서 이뤄지는 연구의 속도와 규모를 고려했을 때 이러한 방법론의 성능은 충분하지 않았다.
공동연구팀은 1,388,606개의 단백질 서열과 이들에게 신뢰성 있게 부여된 EC 번호를 담고 있는 바이오 빅데이터에 딥러닝 기술을 적용해 EC 번호를 빠르고 정확하게 예측할 수 있는 DeepEC를 개발했다.
DeepEC는 주어진 단백질 서열의 EC 번호를 예측하기 위해서 3개의 합성곱 신경망(Convolutional neural network)을 주요 예측기술로 사용하며, 합성곱 신경망으로 EC 번호를 예측하지 못했을 경우 서열정렬(sequence alignment)을 통해서 EC 번호를 예측한다.
연구팀은 더 나아가 단백질 서열의 도메인(domain)과 기질 결합 부위 잔기(binding site residue)에 변이를 인위적으로 주었을 때, DeepEC가 가장 민감하게 해당 변이의 영향을 감지하는 것을 확인했다.
김현욱 교수는 “DeepEC의 성능을 평가하기 위해서 이전에 발표된 5개의 대표적인 EC 번호 예측 방법론과 비교해보니 DeepEC가 가장 빠르고 정확하게 주어진 단백질의 EC 번호를 예측하는 것으로 나타났다”라며 “효소 기능 연구에 크게 이바지할 것으로 기대한다”라고 말했다.
이상엽 특훈교수는 “이번에 개발한 DeepEC를 통해서 지속해서 재생되는 게놈 및 메타 게놈에 존재하는 방대한 효소 단백질 서열의 기능을 보다 효율적이고 정확하게 알아내는 것이 가능해졌다”라고 말했다.
이번 연구는 과학기술정보통신부가 지원하는 기후변화대응기술개발사업의 바이오리파이너리를 위한 시스템대사공학 원천기술개발 과제 및 바이오·의료기술 개발 Korea Bio Grand Challenge 사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 인공지능 기반의 DeepEC를 이용한 효소 기능 EC 번호 예측
2019.07.03 조회수 25531
이상엽 특훈교수, 김현욱 교수, 인공지능 이용한 효소기능 예측 기술 개발
우리 대학 생명화학공학과 이상엽 특훈교수와 김현욱 교수의 초세대 협업연구실 공동연구팀이 딥러닝(deep learning) 기술을 이용해 효소의 기능을 신속하고 정확하게 예측할 수 있는 컴퓨터 방법론 DeepEC를 개발했다.
공동연구팀의 류재용 박사가 1 저자로 참여한 이번 연구결과는 국제학술지 ‘미국 국립과학원 회보(PNAS)’ 6월 20일 자 온라인판에 게재됐다. (논문명 : Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers)
효소는 세포 내의 생화학반응들을 촉진하는 단백질 촉매로 이들의 기능을 정확히 이해하는 것은 세포의 대사(metabolism) 과정을 이해하는 데에 매우 중요하다.
특히 효소들은 다양한 질병 발생 원리 및 산업 생명공학과 밀접한 연관이 있어 방대한 게놈 정보에서 효소들의 기능을 빠르고 정확하게 예측하는 기술은 응용기술 측면에서도 중요하다.
효소의 기능을 표기하는 시스템 중 대표적인 것이 EC 번호(enzyme commission number)이다. EC 번호는 ‘EC 3.4.11.4’처럼 효소가 매개하는 생화학반응들의 종류에 따라 총 4개의 숫자로 구성돼 있다.
중요한 것은 특정 효소에 주어진 EC 번호를 통해서 해당 효소가 어떠한 종류의 생화학반응을 매개하는지 알 수 있다는 것이다. 따라서 게놈으로부터 얻을 수 있는 효소 단백질 서열의 EC 번호를 빠르고 정확하게 예측할 수 있는 기술은 효소 및 대사 관련 문제를 해결하는 데 중요한 역할을 한다.
작년까지 여러 해에 걸쳐 EC 번호를 예측해주는 컴퓨터 방법론들이 최소 10개 이상 개발됐다. 그러나 이들 모두 예측 속도, 예측 정확성 및 예측 가능 범위 측면에서 발전 필요성이 있었다. 특히 현대 생명과학 및 생명공학에서 이뤄지는 연구의 속도와 규모를 고려했을 때 이러한 방법론의 성능은 충분하지 않았다.
공동연구팀은 1,388,606개의 단백질 서열과 이들에게 신뢰성 있게 부여된 EC 번호를 담고 있는 바이오 빅데이터에 딥러닝 기술을 적용해 EC 번호를 빠르고 정확하게 예측할 수 있는 DeepEC를 개발했다.
DeepEC는 주어진 단백질 서열의 EC 번호를 예측하기 위해서 3개의 합성곱 신경망(Convolutional neural network)을 주요 예측기술로 사용하며, 합성곱 신경망으로 EC 번호를 예측하지 못했을 경우 서열정렬(sequence alignment)을 통해서 EC 번호를 예측한다.
연구팀은 더 나아가 단백질 서열의 도메인(domain)과 기질 결합 부위 잔기(binding site residue)에 변이를 인위적으로 주었을 때, DeepEC가 가장 민감하게 해당 변이의 영향을 감지하는 것을 확인했다.
김현욱 교수는 “DeepEC의 성능을 평가하기 위해서 이전에 발표된 5개의 대표적인 EC 번호 예측 방법론과 비교해보니 DeepEC가 가장 빠르고 정확하게 주어진 단백질의 EC 번호를 예측하는 것으로 나타났다”라며 “효소 기능 연구에 크게 이바지할 것으로 기대한다”라고 말했다.
이상엽 특훈교수는 “이번에 개발한 DeepEC를 통해서 지속해서 재생되는 게놈 및 메타 게놈에 존재하는 방대한 효소 단백질 서열의 기능을 보다 효율적이고 정확하게 알아내는 것이 가능해졌다”라고 말했다.
이번 연구는 과학기술정보통신부가 지원하는 기후변화대응기술개발사업의 바이오리파이너리를 위한 시스템대사공학 원천기술개발 과제 및 바이오·의료기술 개발 Korea Bio Grand Challenge 사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 인공지능 기반의 DeepEC를 이용한 효소 기능 EC 번호 예측
2019.07.03 조회수 25531