%EC%9C%A0%EC%A0%84%EC%9E%90+%EB%8F%8C%EC%97%B0%EB%B3%80%EC%9D%B4
-
 3개 학과 공동연구팀, 다학제적 접근 통해 뇌전증 발병 기전 규명
우리 대학 의과학대학원 이정호 교수, 바이오및뇌공학과 백세범 교수, 생명과학과 손종우 교수 공동 연구팀이 MTOR 유전자 돌연변이에 의해 약물 저항성이 높은 뇌전증이 발병하는 메커니즘을 규명했다고 25일 밝혔다.
이번 연구 결과는 극소수의 신경세포에 발생한 돌연변이가 신경망의 과다 활동(hyperactivity) 상태로 이어지는 구체적인 메커니즘을 밝혀, 뇌전증의 발병 원인 및 치료법 개발에 대한 새로운 시각을 제공한다.
특히 3개 학과간 공동 연구팀의 다학제적인 접근을 통해 세포 내 유전학적인 관점에서부터 단일 신경세포의 전기생리학, 이로부터 근접한 거리에 있는 뇌조직의 네트워크, 그리고 뇌 전체 수준에서의 신경망 수준으로 이어지는 다양한 실험 및 시뮬레이션 연구가 이루어져, 뇌전증의 복잡한 발병 메커니즘을 전반적으로 설명하는 성과를 얻었다.
국소피질 이형성증은 대뇌발달 과정에서 일부 신경줄기세포의 mTOR 경로상의 체성유전변이(MTOR, TSC, DEPDC5) 로 발생하는 질환으로, 흔한 뇌전증의 원인 중 하나이며 항뇌전증제 약물 치료에 잘 반응하지 않아 치료가 어렵다. 이에 연구팀은 국소피질 이형성증 환자의 실제 조직과 같은 질환을 가진 동물 모델을 이용한 실험을 통해, 개별 신경세포의 체성유전변이가 신경망 수준의 발작도로 이어지는 구체적인 원리를 규명했다.
먼저 연구팀은 이러한 체성유전변이는 뇌 조직의 5% 이하인 적은 수의 신경세포에서 발생하며, 해당 신경세포들의 전기적 성질이 정상 세포와는 다르게 변화하는 것을 발견했다. 하지만 대다수 정상 세포를 포함한 전반적인 신경망 활동의 시뮬레이션 결과, 이러한 돌연변이는 매우 적은 비율의 신경세포에만 국한돼 있어, 이 세포들 자체의 전기적 성질 변화만으로는 전체 신경망의 비정상적인 활동으로 이어지지 않았고, 이로 인해 뇌전증에서 보이는 신경망 수준의 발작 활성도가 발생하는 이유를 설명할 수 없었다.
이에 연구팀은 후속 실험을 통해, 뇌전증 발작을 유도할 수 있는 활성도가 MTOR 체성 유전변이를 가진 신경세포가 아니라 그 세포들 주변의 변이가 없는 신경세포에 의해 발생하는 것을 발견했다. 이는 유전자 변이를 가진 신경세포의 활성도가 뇌전증의 직접적인 원인이 되는 것이 아니라, 이들 세포가 주변 대다수 비변이 신경세포에 특정 변화를 유도하고 이로 인해 전체 신경망 수준의 발작 활성도가 발생한다는 뜻으로, 뇌 체성유전변이로 인한 비세포 자율성 활성도(non-cell autonomous hyperexcitability)를 보여주는 한 예가 된다.
이에 착안해 추가적인 동물실험과 수술 후 환자 뇌 조직을 이용한 연구를 통해 MTOR 체성유전변이를 가진 세포에서는 ADK(adenosine kinase, 아데노신 키나제) 유전자가 과발현되는 것을 발견했다. 또한, 이로부터 주변 대다수 비변이 신경세포의 네트워크 체계가 교란돼 과활성도가 유도되고, 더 나아가 전체 신경망 수준의 과다 활동으로까지 이어지는 것을 확인했다.
의과학대학원 고현용 박사, 바이오및뇌공학과 장재선 박사, 생명과학과 주상현 학생이 공동 제1 저자로 참여한 이번 연구는 신경학 분야의 국제 학술지 `애널스 오브 뉴롤로지 (Annals of Neurology)' 7월 29일 字에 게재됐다. (논문명: Non-cell autonomous epileptogenesis in focal cortical dysplasia)
이정호, 백세범, 손종우 교수는 "약물 저항성이 높아 기존에 효과적으로 대처할 수 없었던 뇌전증의 발병 원인에 대해 한층 더 깊은 통찰을 제공하는 연구ˮ라며 "한 분야의 실험이나 연구 기법만으로는 해결하기 어려운 문제에 대해, 유전체학, 신경생물학, 계산뇌과학에 걸친 다학제적 접근으로 해결책을 제시한 효과적인 공동연구의 좋은 예시였다ˮ라고 언급했다.
한편 이번 연구는 한국연구재단 이공분야기초연구사업의 리더연구자지원사업 및 중견연구자지원사업, 보건복지부의 질환극복기술개발사업, 서경배 과학재단, 그리고 소바젠의 지원을 받아 수행됐다.
2021.08.26 조회수 13468
3개 학과 공동연구팀, 다학제적 접근 통해 뇌전증 발병 기전 규명
우리 대학 의과학대학원 이정호 교수, 바이오및뇌공학과 백세범 교수, 생명과학과 손종우 교수 공동 연구팀이 MTOR 유전자 돌연변이에 의해 약물 저항성이 높은 뇌전증이 발병하는 메커니즘을 규명했다고 25일 밝혔다.
이번 연구 결과는 극소수의 신경세포에 발생한 돌연변이가 신경망의 과다 활동(hyperactivity) 상태로 이어지는 구체적인 메커니즘을 밝혀, 뇌전증의 발병 원인 및 치료법 개발에 대한 새로운 시각을 제공한다.
특히 3개 학과간 공동 연구팀의 다학제적인 접근을 통해 세포 내 유전학적인 관점에서부터 단일 신경세포의 전기생리학, 이로부터 근접한 거리에 있는 뇌조직의 네트워크, 그리고 뇌 전체 수준에서의 신경망 수준으로 이어지는 다양한 실험 및 시뮬레이션 연구가 이루어져, 뇌전증의 복잡한 발병 메커니즘을 전반적으로 설명하는 성과를 얻었다.
국소피질 이형성증은 대뇌발달 과정에서 일부 신경줄기세포의 mTOR 경로상의 체성유전변이(MTOR, TSC, DEPDC5) 로 발생하는 질환으로, 흔한 뇌전증의 원인 중 하나이며 항뇌전증제 약물 치료에 잘 반응하지 않아 치료가 어렵다. 이에 연구팀은 국소피질 이형성증 환자의 실제 조직과 같은 질환을 가진 동물 모델을 이용한 실험을 통해, 개별 신경세포의 체성유전변이가 신경망 수준의 발작도로 이어지는 구체적인 원리를 규명했다.
먼저 연구팀은 이러한 체성유전변이는 뇌 조직의 5% 이하인 적은 수의 신경세포에서 발생하며, 해당 신경세포들의 전기적 성질이 정상 세포와는 다르게 변화하는 것을 발견했다. 하지만 대다수 정상 세포를 포함한 전반적인 신경망 활동의 시뮬레이션 결과, 이러한 돌연변이는 매우 적은 비율의 신경세포에만 국한돼 있어, 이 세포들 자체의 전기적 성질 변화만으로는 전체 신경망의 비정상적인 활동으로 이어지지 않았고, 이로 인해 뇌전증에서 보이는 신경망 수준의 발작 활성도가 발생하는 이유를 설명할 수 없었다.
이에 연구팀은 후속 실험을 통해, 뇌전증 발작을 유도할 수 있는 활성도가 MTOR 체성 유전변이를 가진 신경세포가 아니라 그 세포들 주변의 변이가 없는 신경세포에 의해 발생하는 것을 발견했다. 이는 유전자 변이를 가진 신경세포의 활성도가 뇌전증의 직접적인 원인이 되는 것이 아니라, 이들 세포가 주변 대다수 비변이 신경세포에 특정 변화를 유도하고 이로 인해 전체 신경망 수준의 발작 활성도가 발생한다는 뜻으로, 뇌 체성유전변이로 인한 비세포 자율성 활성도(non-cell autonomous hyperexcitability)를 보여주는 한 예가 된다.
이에 착안해 추가적인 동물실험과 수술 후 환자 뇌 조직을 이용한 연구를 통해 MTOR 체성유전변이를 가진 세포에서는 ADK(adenosine kinase, 아데노신 키나제) 유전자가 과발현되는 것을 발견했다. 또한, 이로부터 주변 대다수 비변이 신경세포의 네트워크 체계가 교란돼 과활성도가 유도되고, 더 나아가 전체 신경망 수준의 과다 활동으로까지 이어지는 것을 확인했다.
의과학대학원 고현용 박사, 바이오및뇌공학과 장재선 박사, 생명과학과 주상현 학생이 공동 제1 저자로 참여한 이번 연구는 신경학 분야의 국제 학술지 `애널스 오브 뉴롤로지 (Annals of Neurology)' 7월 29일 字에 게재됐다. (논문명: Non-cell autonomous epileptogenesis in focal cortical dysplasia)
이정호, 백세범, 손종우 교수는 "약물 저항성이 높아 기존에 효과적으로 대처할 수 없었던 뇌전증의 발병 원인에 대해 한층 더 깊은 통찰을 제공하는 연구ˮ라며 "한 분야의 실험이나 연구 기법만으로는 해결하기 어려운 문제에 대해, 유전체학, 신경생물학, 계산뇌과학에 걸친 다학제적 접근으로 해결책을 제시한 효과적인 공동연구의 좋은 예시였다ˮ라고 언급했다.
한편 이번 연구는 한국연구재단 이공분야기초연구사업의 리더연구자지원사업 및 중견연구자지원사업, 보건복지부의 질환극복기술개발사업, 서경배 과학재단, 그리고 소바젠의 지원을 받아 수행됐다.
2021.08.26 조회수 13468 -
 유전자 가위를 이용한 새로운 유전자 돌연변이 검출 기술 개발
우리 대학 생명화학공학과 박현규 교수 연구팀이 유전자 가위로 불리는 *크리스퍼(CRISPR-Cas9) 시스템에 의해서 구동되는 *EXPAR 반응을 이용해 유전자 돌연변이를 검출하는 신기술을 개발했다고 11일 밝혔다.
☞ 크리스퍼 (CRISPR-Cas9): 유전자 편집 기술로 DNA를 가위로 자르듯이 특정 부위를 자를 수 있으며, 가이드 RNA(guideRNA)와 Cas9 단백질로 구성된다. 안내자 역할을 하는 guideRNA가 특정 유전자의 위치를 찾아가는 역할을 하고, Cas9 단백질이 유전자를 잘라내는 가위 역할을 한다.
☞ EXPAR: 엑스파(Exponential amplification reaction, EXPAR) 기술은 약 30분의 짧은 반응 시간 내 최대 1억(108)배의 표적 핵산 증폭 효율을 구현함으로써, 높은 활용 가능성을 보유한 기술이다. 구체적으로, EXPAR 기술은 절단 효소 인식 염기서열(템플릿의 중심)과 표적 핵산 상보 염기서열(템플릿의 양 말단)이 수식된 템플릿과 표적 핵산의 혼성화 반응 후, 절단 효소와 DNA 중합 효소의 작용으로 인해 이중가닥 DNA 산물이 지수함수적으로 증폭되는 기술이다.
우리 대학 생명화학공학과 송자연, 김수현 박사가 공동 제1 저자로 참여한 이번 연구는 영국왕립화학회가 발행하는 국제학술지 `나노스케일 (Nanoscale)'에 2021년도 15호 표지(Back cover) 논문으로 지난달 14일 선정됐다. (논문명: A novel method to detect mutation in DNA by utilizing exponential amplification reaction triggered by the CRISPR-Cas9 system)
일반적으로 유전자 돌연변이를 검출하기 위해 중합 효소 연쇄 반응(PCR)을 이용한다. 하지만, 현재까지 개발된 유전자 돌연변이 검출기술들은 낮은 특이도, 낮은 검출 성능, 복잡한 검출 방법, 긴 검출 시간 등의 단점들을 지니고 있다.
연구팀은 이러한 현행 기술의 한계를 극복하기 위해서, 크리스퍼 (CRISPR-Cas9) 시스템을 활용해 검출 특이도를 높이고 EXPAR 등온 증폭 반응을 통해 검출 민감도를 크게 향상시켜서 표적 유전자 돌연변이를 고감도로(검출 한계: 437 aM (아토몰라, Attomolar)) 30분 이내에 검출하는 데 성공했다. 이는 기존 기술 대비 증폭효율 약 10만 배 증가, 검출 시간 약 50% 감소에 해당하는 수치다.
연구팀은 2개의 Cas9/sgRNA 복합체로 구성된 크리스퍼(CRISPR-Cas9) 시스템으로 유전자 돌연변이의 양 끝단을 절단했다. 절단된 짧은 이중 나선 유전자 돌연변이가 EXPAR 반응을 구동시키고 EXPAR 반응 생성물을 통해서 형광 신호가 발생하도록 설계함으로써 표적 유전자 돌연변이를 고감도로 매우 정확하게 검출했다.
연구팀은 이 기술을 통해서, 염색체 DNA 내 HER2와 EGFR 유전자 돌연변이를 성공적으로 검출할 수 있었다. 이러한 유전자 돌연변이는 유방암 및 폐암의 발생에 관여할 뿐만 아니라 특정 치료 약제에 대한 반응을 예측하기 위해서 대표적으로 활용되는 중요한 바이오 마커다.
박현규 교수는 "이번 기술은 CRISPR-Cas9 시스템에 크리스퍼 (CRISPR-Cas9) 시스템에 의해서 구동되는 EXPAR 반응을 이용하여 암 등 다양한 질병에 관여되는 유전자 돌연변이를 고감도로 검출함으로써, 다양한 질병을 조기 진단하고 환자 맞춤형 치료를 구현하는 데 크게 활용될 수 있다ˮ라고 이번 연구의 의의를 설명했다.
한편 이번 연구는 한국연구재단의 지원을 받아 중견연구자지원사업과 글로벌 프런티어지원사업의 일환으로 수행됐다.
2021.05.11 조회수 33674
유전자 가위를 이용한 새로운 유전자 돌연변이 검출 기술 개발
우리 대학 생명화학공학과 박현규 교수 연구팀이 유전자 가위로 불리는 *크리스퍼(CRISPR-Cas9) 시스템에 의해서 구동되는 *EXPAR 반응을 이용해 유전자 돌연변이를 검출하는 신기술을 개발했다고 11일 밝혔다.
☞ 크리스퍼 (CRISPR-Cas9): 유전자 편집 기술로 DNA를 가위로 자르듯이 특정 부위를 자를 수 있으며, 가이드 RNA(guideRNA)와 Cas9 단백질로 구성된다. 안내자 역할을 하는 guideRNA가 특정 유전자의 위치를 찾아가는 역할을 하고, Cas9 단백질이 유전자를 잘라내는 가위 역할을 한다.
☞ EXPAR: 엑스파(Exponential amplification reaction, EXPAR) 기술은 약 30분의 짧은 반응 시간 내 최대 1억(108)배의 표적 핵산 증폭 효율을 구현함으로써, 높은 활용 가능성을 보유한 기술이다. 구체적으로, EXPAR 기술은 절단 효소 인식 염기서열(템플릿의 중심)과 표적 핵산 상보 염기서열(템플릿의 양 말단)이 수식된 템플릿과 표적 핵산의 혼성화 반응 후, 절단 효소와 DNA 중합 효소의 작용으로 인해 이중가닥 DNA 산물이 지수함수적으로 증폭되는 기술이다.
우리 대학 생명화학공학과 송자연, 김수현 박사가 공동 제1 저자로 참여한 이번 연구는 영국왕립화학회가 발행하는 국제학술지 `나노스케일 (Nanoscale)'에 2021년도 15호 표지(Back cover) 논문으로 지난달 14일 선정됐다. (논문명: A novel method to detect mutation in DNA by utilizing exponential amplification reaction triggered by the CRISPR-Cas9 system)
일반적으로 유전자 돌연변이를 검출하기 위해 중합 효소 연쇄 반응(PCR)을 이용한다. 하지만, 현재까지 개발된 유전자 돌연변이 검출기술들은 낮은 특이도, 낮은 검출 성능, 복잡한 검출 방법, 긴 검출 시간 등의 단점들을 지니고 있다.
연구팀은 이러한 현행 기술의 한계를 극복하기 위해서, 크리스퍼 (CRISPR-Cas9) 시스템을 활용해 검출 특이도를 높이고 EXPAR 등온 증폭 반응을 통해 검출 민감도를 크게 향상시켜서 표적 유전자 돌연변이를 고감도로(검출 한계: 437 aM (아토몰라, Attomolar)) 30분 이내에 검출하는 데 성공했다. 이는 기존 기술 대비 증폭효율 약 10만 배 증가, 검출 시간 약 50% 감소에 해당하는 수치다.
연구팀은 2개의 Cas9/sgRNA 복합체로 구성된 크리스퍼(CRISPR-Cas9) 시스템으로 유전자 돌연변이의 양 끝단을 절단했다. 절단된 짧은 이중 나선 유전자 돌연변이가 EXPAR 반응을 구동시키고 EXPAR 반응 생성물을 통해서 형광 신호가 발생하도록 설계함으로써 표적 유전자 돌연변이를 고감도로 매우 정확하게 검출했다.
연구팀은 이 기술을 통해서, 염색체 DNA 내 HER2와 EGFR 유전자 돌연변이를 성공적으로 검출할 수 있었다. 이러한 유전자 돌연변이는 유방암 및 폐암의 발생에 관여할 뿐만 아니라 특정 치료 약제에 대한 반응을 예측하기 위해서 대표적으로 활용되는 중요한 바이오 마커다.
박현규 교수는 "이번 기술은 CRISPR-Cas9 시스템에 크리스퍼 (CRISPR-Cas9) 시스템에 의해서 구동되는 EXPAR 반응을 이용하여 암 등 다양한 질병에 관여되는 유전자 돌연변이를 고감도로 검출함으로써, 다양한 질병을 조기 진단하고 환자 맞춤형 치료를 구현하는 데 크게 활용될 수 있다ˮ라고 이번 연구의 의의를 설명했다.
한편 이번 연구는 한국연구재단의 지원을 받아 중견연구자지원사업과 글로벌 프런티어지원사업의 일환으로 수행됐다.
2021.05.11 조회수 33674 -
 조광현 교수, 암세포 유형별 최적 약물표적 발굴기술 개발
〈 최민수 박사, 조광현 교수 〉
우리 대학 바이오및뇌공학과 조광현 교수 연구팀이 암세포의 유형에 따라 최적의 약물 표적을 찾는 기술을 개발했다.
이는 시스템생물학을 이용해 암세포의 유전자변이가 반영된 분자네트워크의 다이나믹스(동역학)를 분석해 약물의 반응을 예측하는 기술로 향후 암 관련 신약 개발에 크게 기여할 것으로 기대된다.
최민수, 시 주 (Shi Jue), 주 양팅 (Zhu Yanting), 양 루젠 (Yang Ruizhen)이 참여한 이번 연구는 ‘네이처 커뮤니케이션즈(Nature Communications)’ 12월 5일자 온라인 판에 게재됐다.
인간의 암세포는 유전자 돌연변이, 유전체 단위의 반복적 변이 등 여러 형태의 유전자 변이가 있다. 이러한 변이는 같은 암종에서도 암세포에 따라 많은 차이를 보이기 때문에 약물에 대한 반응도 다양하다.
암 연구자들은 암 환자에게서 빈번하게 발견되는 유전자변이를 파악하고 이 중 특정 약물의 지표로 사용될 수 있는 유전자변이를 찾기 위해 노력해 왔다. 이러한 연구는 단일 유전자변이의 발견 또는 유전자네트워크의 구조적 특징 분석에 초점이 맞춰져 있다.
하지만 이러한 접근 방법은 암세포 내 다양한 유전자 및 단백질의 상호작용에 의해 유발되는 암의 생물학적 특성과 이로 인한 약물반응의 차이를 설명하지 못하는 한계가 있다.
암세포의 유전자변이는 해당 유전자 기능 뿐 아니라 이 유전자와 상호작용하는 다른 유전자, 단백질에 영향을 미치기 때문에 결과적으로 분자네트워크의 다이나믹스(동역학) 특성에 변화를 일으킨다.
이로 인해 항암제에 대한 암세포의 반응이 변화하게 된다. 따라서 분자네트워크의 다이나믹스(동역학) 특성을 무시하고 소수의 암 관련 유전자를 표적으로 하는 현재의 치료법은 일부 환자에게만 유용하고 약물저항성을 갖는 대다수 환자에게는 효과적으로 적용되지 못한다.
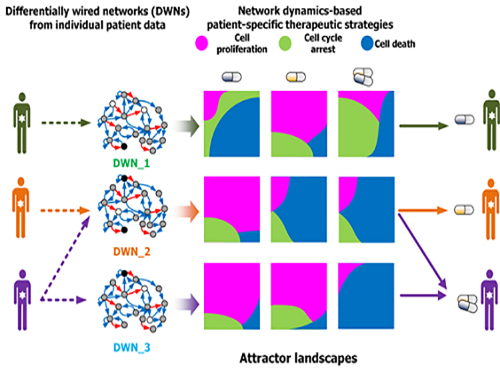
조 교수 연구팀은 문제 해결을 위해 슈퍼컴퓨팅을 이용한 대규모 컴퓨터시뮬레이션과 세포 실험을 융합해 암세포 분자네트워크의 다이나믹스(동역학) 변화를 분석했다.
이를 통해 약물반응을 예측해 유형별 암세포의 최적 약물 표적을 발굴하는 기술을 개발했다. 이 기술은 대다수 암 발생에 관여하는 것으로 알려진 암 억제 유전자 p53의 분자조절네트워크에 시범적으로 적용됐다.
연구팀은 국제 컨소시엄인 암 세포주 백과사전(CCLE : The Cancer Cell Line Encyclopedia)에 공개된 대규모 암세포 유전체 데이터를 분자네트워크에 반영해 구축했으며 유전변이의 특성에 따라 서로 다른 분자네트워크를 생성했다.
각 분자네트워크에 대해 약물반응을 모사한 섭동분석을 수행해 약물반응을 나타내는 암세포의 변화를 정량화하고 군집화했다. 그 후 컴퓨터시뮬레이션 분석을 통해 효능, 조합에 따른 시너지효과 등 약물반응정도를 예측했다.
이러한 컴퓨터시뮬레이션 결과를 토대로 폐암, 유방암, 골종양, 피부암, 신장암, 난소암 등 다양한 암세포주를 대상으로 약물반응 실험을 수행해 비교 검증했다.
이 기술은 임의의 분자네트워크에 대해서 동일한 방식으로 적용할 수 있고 최적의 약물 표적을 발굴해 개인 맞춤치료에 활용가능하다.
연구팀은 암세포의 이질성에 따른 다양한 약물반응의 원인을 특정 유전자나 단백질뿐만 아니라 상호조절작용을 종합적으로 고려해 분석할 수 있게 됐다고 밝혔다.
또한 약물저항성의 원인을 사전에 예측하고 이를 억제할 수 있는 최적의 약물 표적을 발굴할 수 있게 됐고 기존 약물의 새로운 적용대상을 찾는 약물재창출에 활용될 수 있는 핵심 원천기술을 확보하게 됐다고 말했다.
조 교수는 “암세포별 유전변이는 약물반응 다양성의 원인이지만 지금까지 이에 대한 총체적 분석이 이뤄지지 못했다”며 “시스템생물학을 통해 암세포 유형별 분자네트워크의 약물반응을 시뮬레이션으로 분석해 약물 반응의 근본적 원리를 파악하고 새로운 개념의 최적 약물 타겟을 발굴할 수 있게 됐다”고 말했다.
이번 연구는 과학기술정보통신부와 한국연구재단의 중견연구자지원사업과 바이오의료기술개발사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 컴퓨터시뮬레이션을 통한 암세포 유형별 약물반응 예측 및 세포실험 비교 검증
그림2. 암세포별 분자네트워크의 동역학 분석에 기반한 약물반응 예측 및 군집화
그림3. 세포 분자네트워크 분석에 따른 암세포 유형별 약물타겟 발굴 및 암환자별 맞춤치료 전략 수립
2017.12.07 조회수 24784
조광현 교수, 암세포 유형별 최적 약물표적 발굴기술 개발
〈 최민수 박사, 조광현 교수 〉
우리 대학 바이오및뇌공학과 조광현 교수 연구팀이 암세포의 유형에 따라 최적의 약물 표적을 찾는 기술을 개발했다.
이는 시스템생물학을 이용해 암세포의 유전자변이가 반영된 분자네트워크의 다이나믹스(동역학)를 분석해 약물의 반응을 예측하는 기술로 향후 암 관련 신약 개발에 크게 기여할 것으로 기대된다.
최민수, 시 주 (Shi Jue), 주 양팅 (Zhu Yanting), 양 루젠 (Yang Ruizhen)이 참여한 이번 연구는 ‘네이처 커뮤니케이션즈(Nature Communications)’ 12월 5일자 온라인 판에 게재됐다.
인간의 암세포는 유전자 돌연변이, 유전체 단위의 반복적 변이 등 여러 형태의 유전자 변이가 있다. 이러한 변이는 같은 암종에서도 암세포에 따라 많은 차이를 보이기 때문에 약물에 대한 반응도 다양하다.
암 연구자들은 암 환자에게서 빈번하게 발견되는 유전자변이를 파악하고 이 중 특정 약물의 지표로 사용될 수 있는 유전자변이를 찾기 위해 노력해 왔다. 이러한 연구는 단일 유전자변이의 발견 또는 유전자네트워크의 구조적 특징 분석에 초점이 맞춰져 있다.
하지만 이러한 접근 방법은 암세포 내 다양한 유전자 및 단백질의 상호작용에 의해 유발되는 암의 생물학적 특성과 이로 인한 약물반응의 차이를 설명하지 못하는 한계가 있다.
암세포의 유전자변이는 해당 유전자 기능 뿐 아니라 이 유전자와 상호작용하는 다른 유전자, 단백질에 영향을 미치기 때문에 결과적으로 분자네트워크의 다이나믹스(동역학) 특성에 변화를 일으킨다.
이로 인해 항암제에 대한 암세포의 반응이 변화하게 된다. 따라서 분자네트워크의 다이나믹스(동역학) 특성을 무시하고 소수의 암 관련 유전자를 표적으로 하는 현재의 치료법은 일부 환자에게만 유용하고 약물저항성을 갖는 대다수 환자에게는 효과적으로 적용되지 못한다.
조 교수 연구팀은 문제 해결을 위해 슈퍼컴퓨팅을 이용한 대규모 컴퓨터시뮬레이션과 세포 실험을 융합해 암세포 분자네트워크의 다이나믹스(동역학) 변화를 분석했다.
이를 통해 약물반응을 예측해 유형별 암세포의 최적 약물 표적을 발굴하는 기술을 개발했다. 이 기술은 대다수 암 발생에 관여하는 것으로 알려진 암 억제 유전자 p53의 분자조절네트워크에 시범적으로 적용됐다.
연구팀은 국제 컨소시엄인 암 세포주 백과사전(CCLE : The Cancer Cell Line Encyclopedia)에 공개된 대규모 암세포 유전체 데이터를 분자네트워크에 반영해 구축했으며 유전변이의 특성에 따라 서로 다른 분자네트워크를 생성했다.
각 분자네트워크에 대해 약물반응을 모사한 섭동분석을 수행해 약물반응을 나타내는 암세포의 변화를 정량화하고 군집화했다. 그 후 컴퓨터시뮬레이션 분석을 통해 효능, 조합에 따른 시너지효과 등 약물반응정도를 예측했다.
이러한 컴퓨터시뮬레이션 결과를 토대로 폐암, 유방암, 골종양, 피부암, 신장암, 난소암 등 다양한 암세포주를 대상으로 약물반응 실험을 수행해 비교 검증했다.
이 기술은 임의의 분자네트워크에 대해서 동일한 방식으로 적용할 수 있고 최적의 약물 표적을 발굴해 개인 맞춤치료에 활용가능하다.
연구팀은 암세포의 이질성에 따른 다양한 약물반응의 원인을 특정 유전자나 단백질뿐만 아니라 상호조절작용을 종합적으로 고려해 분석할 수 있게 됐다고 밝혔다.
또한 약물저항성의 원인을 사전에 예측하고 이를 억제할 수 있는 최적의 약물 표적을 발굴할 수 있게 됐고 기존 약물의 새로운 적용대상을 찾는 약물재창출에 활용될 수 있는 핵심 원천기술을 확보하게 됐다고 말했다.
조 교수는 “암세포별 유전변이는 약물반응 다양성의 원인이지만 지금까지 이에 대한 총체적 분석이 이뤄지지 못했다”며 “시스템생물학을 통해 암세포 유형별 분자네트워크의 약물반응을 시뮬레이션으로 분석해 약물 반응의 근본적 원리를 파악하고 새로운 개념의 최적 약물 타겟을 발굴할 수 있게 됐다”고 말했다.
이번 연구는 과학기술정보통신부와 한국연구재단의 중견연구자지원사업과 바이오의료기술개발사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 컴퓨터시뮬레이션을 통한 암세포 유형별 약물반응 예측 및 세포실험 비교 검증
그림2. 암세포별 분자네트워크의 동역학 분석에 기반한 약물반응 예측 및 군집화
그림3. 세포 분자네트워크 분석에 따른 암세포 유형별 약물타겟 발굴 및 암환자별 맞춤치료 전략 수립
2017.12.07 조회수 24784 -
 조광현 교수, 대장암 유발하는 돌연변이 유전자의 네트워크 원리 규명
〈 왼쪽위부터 시계방향으로 이종훈 박사과정, 공정렬 박사과정, 조광현 교수, 신동관 연구교수 〉
우리 대학 바이오및뇌공학과 조광현 교수 연구팀이 대장암이 발병하는 과정에서 생기는 유전자 네트워크의 원리를 규명하는 데 성공했다.
이를 통해 대장암의 근본적인 발병 원리를 밝혀낼 뿐 아니라 향후 새로운 개념의 효과적인 항암제의 분자표적을 찾는데 활용될 것으로 기대된다. 또한 4차 산업혁명의 핵심 기술로 주목받는 IT와 BT의 융합연구인 시스템생물학 연구로 규명해냈다는 의의를 갖는다.
신동관 박사, 이종훈, 공정렬 학생연구원 등이 함께 참여한 이번 연구는 ‘네이처 커뮤니케이션즈(Nature Communications)’ 2일자 온라인 판에 게재됐다.
인간의 암은 유전자 돌연변이에 의해 발생한다. 이 돌연변이의 빈도는 암종에 따라 차이가 나는데 백혈병, 소아암은 10여 개 정도이지만 성인 고형암은 평균 50여 개, 폐암 등의 외부인자로 인한 경우는 수백 개에 이른다.
전 세계 암연구자들은 암 치료를 위해 환자들에게서 빈번하게 발견되는 유전자 돌연변이들을 파악하고 이 중 주요 암 유발 유전자를 찾아내 표적 항암제를 개발하고자 노력했다.
그러나 유전자 돌연변이는 해당 유전자의 기능에만 영향을 주는 게 아니라 그 유전자와 상호작용하는 다른 유전자에게도 영향을 끼친다. 따라서 이러한 유전자 네트워크의 원리를 모른 채 소수의 암 유발 유전자를 대상으로 하는 현재의 치료법은 일부에게만 효과가 있고 쉽게 약물의 내성을 일으키는 한계가 있다.
조 교수 연구팀은 대장암 환자의 대규모 유전체 데이터를 이용해 유전자 상호작용 네트워크에서 나타나는 다중 돌연변이의 협력적 효과에 대한 수학모형을 구축했다.
이는 국제 암유전체컨소시엄에서 발표한 전암 유전체데이터베이스(TCGA: The Cancer Genome Atlas)를 토대로 구축한 것으로, 유전자 네트워크에서 나타나는 돌연변이의 영향력을 정량화하고 이를 이용해 대장암 환자 군을 임상 특징에 따라 군집화 하는데 성공했다.
또한 대규모 컴퓨터 시뮬레이션 분석을 통해 암 발생 과정에서 나타나는 임계전이(critical transition) 현상을 밝혀내 숨겨진 유전자 네트워크의 원리를 최초로 규명했다.
임계전이란 상전이와 같이 물질의 상태가 갑작스럽게 변화하는 현상을 말한다. 암 발생 과정에서는 유전자 돌연변이의 발생 순서를 추적하기 어렵기 때문에 전이 현상이 존재하는지 확인할 수 없었다.
연구팀은 시스템생물학 기반의 연구방법을 이용해 확인한 결과 기존의 대장암에서 잘 알려진 암 유발 유전자 돌연변이의 발생 순서를 따르는 경우에 임계전이 현상을 보임을 발견했다.
이번에 개발한 수학모형을 활용하면 암환자에게 발생하는 다수 유전자 돌연변이의 영향을 가장 효과적으로 저해할 수 있는 새로운 항암 표적 약물이 개발될 것으로 기대된다.
특히 주요 암 유발 유전자 뿐 아니라 돌연변이의 영향을 받는 다른 모든 유전자들을 대상으로 종합적으로 평가해 효과적인 약물 표적을 찾아낼 수 있다.
조 교수는 “지금껏 다수 유전자들의 돌연변이가 암 발생에 어떻게 기여하는지 밝혀진 바가 없었다”며 “이번 연구에서는 시스템생물학으로 암세포의 발달과정에서 유전자 네트워크의 원리를 최초로 밝힘으로써 새로운 차원의 항암제 표적을 발굴할 수 있는 가능성을 제시했다”고 말했다.
이번 연구는 과학기술정보통신부와 한국연구재단의 중견연구자지원사업과 바이오의료기술개발사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 유전자 돌연변이의 영향력 전파에 의한 거대 클러스터의 형성
그림2. 암발생 과정에서 돌연변이 협력효과의 임계전이 현상
2017.11.07 조회수 25217
조광현 교수, 대장암 유발하는 돌연변이 유전자의 네트워크 원리 규명
〈 왼쪽위부터 시계방향으로 이종훈 박사과정, 공정렬 박사과정, 조광현 교수, 신동관 연구교수 〉
우리 대학 바이오및뇌공학과 조광현 교수 연구팀이 대장암이 발병하는 과정에서 생기는 유전자 네트워크의 원리를 규명하는 데 성공했다.
이를 통해 대장암의 근본적인 발병 원리를 밝혀낼 뿐 아니라 향후 새로운 개념의 효과적인 항암제의 분자표적을 찾는데 활용될 것으로 기대된다. 또한 4차 산업혁명의 핵심 기술로 주목받는 IT와 BT의 융합연구인 시스템생물학 연구로 규명해냈다는 의의를 갖는다.
신동관 박사, 이종훈, 공정렬 학생연구원 등이 함께 참여한 이번 연구는 ‘네이처 커뮤니케이션즈(Nature Communications)’ 2일자 온라인 판에 게재됐다.
인간의 암은 유전자 돌연변이에 의해 발생한다. 이 돌연변이의 빈도는 암종에 따라 차이가 나는데 백혈병, 소아암은 10여 개 정도이지만 성인 고형암은 평균 50여 개, 폐암 등의 외부인자로 인한 경우는 수백 개에 이른다.
전 세계 암연구자들은 암 치료를 위해 환자들에게서 빈번하게 발견되는 유전자 돌연변이들을 파악하고 이 중 주요 암 유발 유전자를 찾아내 표적 항암제를 개발하고자 노력했다.
그러나 유전자 돌연변이는 해당 유전자의 기능에만 영향을 주는 게 아니라 그 유전자와 상호작용하는 다른 유전자에게도 영향을 끼친다. 따라서 이러한 유전자 네트워크의 원리를 모른 채 소수의 암 유발 유전자를 대상으로 하는 현재의 치료법은 일부에게만 효과가 있고 쉽게 약물의 내성을 일으키는 한계가 있다.
조 교수 연구팀은 대장암 환자의 대규모 유전체 데이터를 이용해 유전자 상호작용 네트워크에서 나타나는 다중 돌연변이의 협력적 효과에 대한 수학모형을 구축했다.
이는 국제 암유전체컨소시엄에서 발표한 전암 유전체데이터베이스(TCGA: The Cancer Genome Atlas)를 토대로 구축한 것으로, 유전자 네트워크에서 나타나는 돌연변이의 영향력을 정량화하고 이를 이용해 대장암 환자 군을 임상 특징에 따라 군집화 하는데 성공했다.
또한 대규모 컴퓨터 시뮬레이션 분석을 통해 암 발생 과정에서 나타나는 임계전이(critical transition) 현상을 밝혀내 숨겨진 유전자 네트워크의 원리를 최초로 규명했다.
임계전이란 상전이와 같이 물질의 상태가 갑작스럽게 변화하는 현상을 말한다. 암 발생 과정에서는 유전자 돌연변이의 발생 순서를 추적하기 어렵기 때문에 전이 현상이 존재하는지 확인할 수 없었다.
연구팀은 시스템생물학 기반의 연구방법을 이용해 확인한 결과 기존의 대장암에서 잘 알려진 암 유발 유전자 돌연변이의 발생 순서를 따르는 경우에 임계전이 현상을 보임을 발견했다.
이번에 개발한 수학모형을 활용하면 암환자에게 발생하는 다수 유전자 돌연변이의 영향을 가장 효과적으로 저해할 수 있는 새로운 항암 표적 약물이 개발될 것으로 기대된다.
특히 주요 암 유발 유전자 뿐 아니라 돌연변이의 영향을 받는 다른 모든 유전자들을 대상으로 종합적으로 평가해 효과적인 약물 표적을 찾아낼 수 있다.
조 교수는 “지금껏 다수 유전자들의 돌연변이가 암 발생에 어떻게 기여하는지 밝혀진 바가 없었다”며 “이번 연구에서는 시스템생물학으로 암세포의 발달과정에서 유전자 네트워크의 원리를 최초로 밝힘으로써 새로운 차원의 항암제 표적을 발굴할 수 있는 가능성을 제시했다”고 말했다.
이번 연구는 과학기술정보통신부와 한국연구재단의 중견연구자지원사업과 바이오의료기술개발사업의 지원을 받아 수행됐다.
□ 그림 설명
그림1. 유전자 돌연변이의 영향력 전파에 의한 거대 클러스터의 형성
그림2. 암발생 과정에서 돌연변이 협력효과의 임계전이 현상
2017.11.07 조회수 25217